高活性纤溶酶菌株筛选与基因克隆表达
2019-09-10王志会谢和
王志会 谢和


摘 要:本研究以本实验室曾在酒曲、酒糟、窖泥等样品中分离的具有酱香味的13株菌株作为出发菌株,经脱脂奶粉平板、纤维蛋白双层平板筛选出纤溶酶活性最高的菌株GZHZV-8,结合形态学特征、生理生化特征及16S rDNA序列同源性分析,GZHZV-8菌株为解淀粉芽孢杆菌(Bacillus amyloliquefaciens)。以GZHZV-8菌株总DNA为模板扩增出纤溶酶基因的编码区,与pMD18-T载体连接、转化至大肠杆菌(Escherichia coli) DH5α,分析表明纤溶酶基因开放阅读框包含1092 bp碱基,编码363个氨基酸。纤溶酶基因与表达载体pET-22b(+)连接转化至E.coli BL21中表达,表达菌株经IPTG诱导表达分泌活性纤溶酶,培养后测得发酵上清液、菌体细胞破碎上清液和菌体细胞破碎沉淀的纤溶酶活力分别为613.26 IU/mL、407.38 IU/mL、993.12 IU/mL。
关键词:纤溶酶; 基因克隆; 序列分析; 表达
中图分类号:Q939.9
文献标识码:A
文章编号:1008-0457(2019)02-0001-07 国际DOI编码:10.15958/j.cnki.sdnyswxb.2019.02.001
Abstract: In this study, 13 Maotai-flavor producing strains isolated from samples such as daqu, fermented grains and pit mud were used as the starting strains. GZHZV-8, the strain with the highest fibrinolytic activity, was screened by the skim milk powder plate and fibrin double layer plate. Combined with morphological characteristics, physiological and biochemical characteristics and 16S rDNA sequence homology analysis, the strain GZHZV-8 was identified as Bacillus amyloliquefaciens. The coding region of the fibrinolytic enzyme gene was amplified by using the total DNA of GZHZV-8 as a template, and ligated with pMD18-T vector and transformed into Esc-herichia coli DH5α. The results indicated that the open reading frame of fibrinolytic enzyme gene contained 1092 bp and encoded 363 amino acids. The fibrinolytic enzyme gene was ligated with expression vector pET-22b(+) and transformed into E. coli BL21, and the expression strain was induced to express the active fibrinolytic enzyme by IPTG. The fibrinolytic activity of fermentation supernatant, cell disruption supernatant and cell debris was 613.26 IU/mL, 407.38 IU/mL and 993.12 IU/mL, respectively.
Key words:fibrinolytic enzyme; gene cloning; sequence analysis; expression
醬香型白酒中微生物种类丰富[1],许多微生物不仅具有产酱香风味物质的功能,还具有体外溶栓、抗凝血的生理作用[2-3]。日本学者须见洋行从日本传统发酵食品纳豆中提取出一种具有纤溶活性的纳豆激酶[4]。傅莉等[5]在1997年报道了一株产纤溶酶的枯草芽孢杆菌(Bacillus subtilis),且在最适宜条件下,酶活力为200 U/mL。Nakamura T等[6]首次对纳豆激酶的基因进行了研究,发现该基因是一种丝氨酸蛋白酶,可直接降解纤维蛋白。AI Hai-xin等[7]构建纳豆激酶原核表达载体在E.coli BL21中克隆高效表达,产量显著提高,但酶活性相对降低。刘北域等[8]将纳豆激酶基因克隆在大肠杆菌-枯草杆菌穿梭表达载体pBNK中,在枯草杆菌中表达,酶比活力达12000 U/mg。Lu Xiao等[9]将来自解淀粉芽孢杆菌的α-淀粉酶基因的启动子和信号肽编码序列克隆并与编码枯草杆菌蛋白酶DFE的前肽和成熟肽的序列融合。将该杂合基因插入大肠杆菌/枯草芽孢杆菌穿梭质粒载体pSUGV4中。重组枯草杆菌蛋白酶DFE基因在枯草芽孢杆菌WB600中成功表达,其纤维蛋白溶解活性为200 IU/mL。王开敏等人[10]筛选出了高纤溶活性菌株,用PCR技术扩增出纤溶酶基因的编码区,将编码区序列连载到穿梭载体pYES2上,再导入到酿酒酵母(Saccharomyces cerevisiae)H158细胞中,经诱导发酵后纤溶酶活力为222.49 IU/mL。本研究以本实验室保存的分离自酒曲、酒糟、窖泥等样品中产酱香细菌中筛选产高纤溶酶活性的菌株为材料结合形态学特征、生理生化特征以及分子生物学方法鉴定此菌株,并利用基因工程技术对纤溶酶基因进行克隆与表达。
1 材料与方法
1.1 菌株
GZJSⅠ-2、GZJSⅠ-5、GZJSⅠ-12、GZJSⅡ-1、GZHZⅡ-8、GZHZⅤ-9、GZHZⅤ-8、GZHZⅥ-11、GZJSⅠ-12-2、GZJSⅠ-12-4、GZJSⅠ-12-5、GZJSⅠ-12-7、GZJSⅠ-12-12菌株为贵州大学微生物实验室保存。大肠杆菌E.coli DH5α、大肠杆菌E.coli BL21购自于昆明硕擎生物。
1.2 试剂
限制性内切酶(BamH I、Hind III)、T4连接酶、PrimeSTAR购自TaKaRa;纤维蛋白原购自索莱宝;凝血酶(840 IU/支)、尿激酶(1240 IU/支)购自中国食品药品检定研究院;pMD18-T购自TaKaRa;pET-22b(+)购自淼灵质粒平台。
1.3 主要培养基
牛肉膏蛋白胨培养基[11],种子培养基、发酵培养基及产纤溶酶初筛培养基[3]。
1.4 方法
1.4.1 产纤溶酶菌株的初筛[3]
将实验菌株活化,并转接到脱脂奶粉初筛培养基上,37℃恒温倒置培养48 h后对菌落直径(d)和透明圈直径(D)各进行3次测定,计算D/d(D/d:菌株产蛋白能力的大小),将产蛋白能力相对较强的菌株定为复筛的出发菌株。
1.4.2 产纤溶酶菌株的复筛[3]
初筛出的菌株接种于种子培养基,30℃、150 r/min震荡培养24 h后按照4%的量转接于发酵培养基中,30℃、150 r/min震荡培养96 h。将发酵液在4℃,8000 r/min离心20 min,取10 μL上清液點样于纤维蛋白双层平板上,室温置10 min后倒置于37℃恒温培养箱培养18 h,测定溶解圈垂直直径大小,各测3次计算平均值,垂直直径的乘积为溶解圈的面积。溶解圈面积最大的菌株作为高活性纤溶酶菌株。
1.4.3 纤溶酶活性测定
根据Astup报道[12]的纤维蛋白双层平板法测定纤溶酶活力,并以尿激酶(标准品)做纤溶酶活力标准曲线。
1.4.4 GZHZV-8菌种鉴定
菌株形态特征及生理生化特征鉴定根据《伯杰手册》(R.E.布坎南和N.E.吉本斯等,1984)第八版和《常见细菌鉴定手册》(东秀珠和蔡妙英,2001)[13-14]进行。
参照天根基因组DNA提取试剂盒提取DNA,利用16S rDNA基因扩增引物[15](27F:5-AGAGTTTGATCMTGGCTCAG-3;1492R:5-TACGGYTACCTTGTTACGACTT-3)进行扩增,PCR扩增体系:DNA模板2 μL,引物各2 μL,2×Es Taq MasterMix 25 μL,ddH2O 19 μL。PCR反应条件:95℃预变性5 min;95℃变性30 s,55℃退火30 s,72℃延伸90 s,循环30次;72℃终延伸10 min。扩增产物经1%琼脂糖电泳分析后由英潍捷基双向测序。16S rDNA基因测序结果与细菌模式菌株网(http://www.ezbiocloud.net/ezgenome)中的序列进行比对,下载相似度较高(>90%)的相关模式菌株16S rDNA,采用邻接法(Neighbor-Joining)利用MEGA 5.0构建系统发育树[15]。
1.4.5 扩增纤溶酶基因
扩增根据NCBI报道的纤溶酶基因DNA序列(Gene Bank AY720895.2)的编码区应用Primer Premier 5.0设计并合成一对引物(F:5-CGGGATCCGTGAGAGGCAAAAAGGTA TGG-3BamH I;R:5-CCCAAGCTTTTACTGAGC TGCCGCCTGTACG-3Hind III)。
以筛选菌株总DNA为模板,进行PCR扩增,PCR扩增体系:5×PrimeSTAR Buffer 10 μL,dNTP Mixture 4 μL,Primer F 1 μL,Primer R 1 μL,Template 1 μL,PrimeSTAR HS DNA Polymerase 0.5 μL,无菌水32.5 μL。PCR反应程序为:98℃预变性3 min,98℃变性10 s,60℃退火10 s,72℃延伸90 s,35个循环,72℃延伸5 min。PCR产物用1%凝胶电泳检测。
1.4.6 纤溶酶基因克隆及重组子的筛选鉴定
根据DNA胶回收试剂盒对PCR产物进行切胶回收纯化,用限制性内切酶BamH I与Hind III对pMD18-T克隆载体与PCR纯化产物进行双酶切再切胶回收纯化、连接、转化至克隆宿主E.coli DH5α感受态细胞中,将一定量的转化液涂布于相应的抗性LB平板上,通过PCR扩增、质粒双酶切及测序验证阳性克隆子。
1.4.7 纤溶酶基因的序列分析[16]
利用NCBI、ExPASy在线工具对纤溶酶基因的序列进行预测分析。
1.4.8 表达载体的构建
用BamH I与Hind III双酶切克隆质粒与表达载体PET-22b(+),T4 DNA Ligase连接,转化至大肠杆菌E.coli BL21,取一定量的转化液涂布于相应的抗性LB平板上,通过PCR扩增、质粒双酶切、测序验证阳性重组菌。
1.4.9 重组菌纤溶酶活性测定
用一定量的IPTG诱导发酵重组菌,在4℃、8000 r/min离心20 min,分别收集上清液和菌体,菌体经超声波破碎(破碎5 s,间隔5 s),于4℃、10000 r/min离心20 min,分别收集破碎上清液与沉淀。测定pET-22b(+)空载体、经IPTG诱导重组菌的发酵上清液、菌体破碎上清液及沉淀的纤溶酶活性。
2 结果与分析
2.1 产纤溶酶菌株的初筛
13株菌均具有不同程度的产蛋白能力,经过脱脂奶粉平板测得13株菌的产蛋白能力(表1)。
2.2 产纤溶酶菌株的复筛
根据初筛时测定的菌株产蛋白能力大小,挑选6株产蛋白能力较强的菌株进行复筛,由表2可知GZHZV-8纤溶酶活力最高,为2344.23 IU/mL。纤维蛋白平板法测定的溶解圈见图1。
2.3 尿激酶标准曲线
按照尿激酶标准曲线制作方法,绘制尿激酶活性标准曲线,标准曲线方程为:y=1.1938x-0.2265,R2为0.9976,具有良好的线性关系(图2)。
2.4 菌种鉴定
2.4.1 形态学鉴定
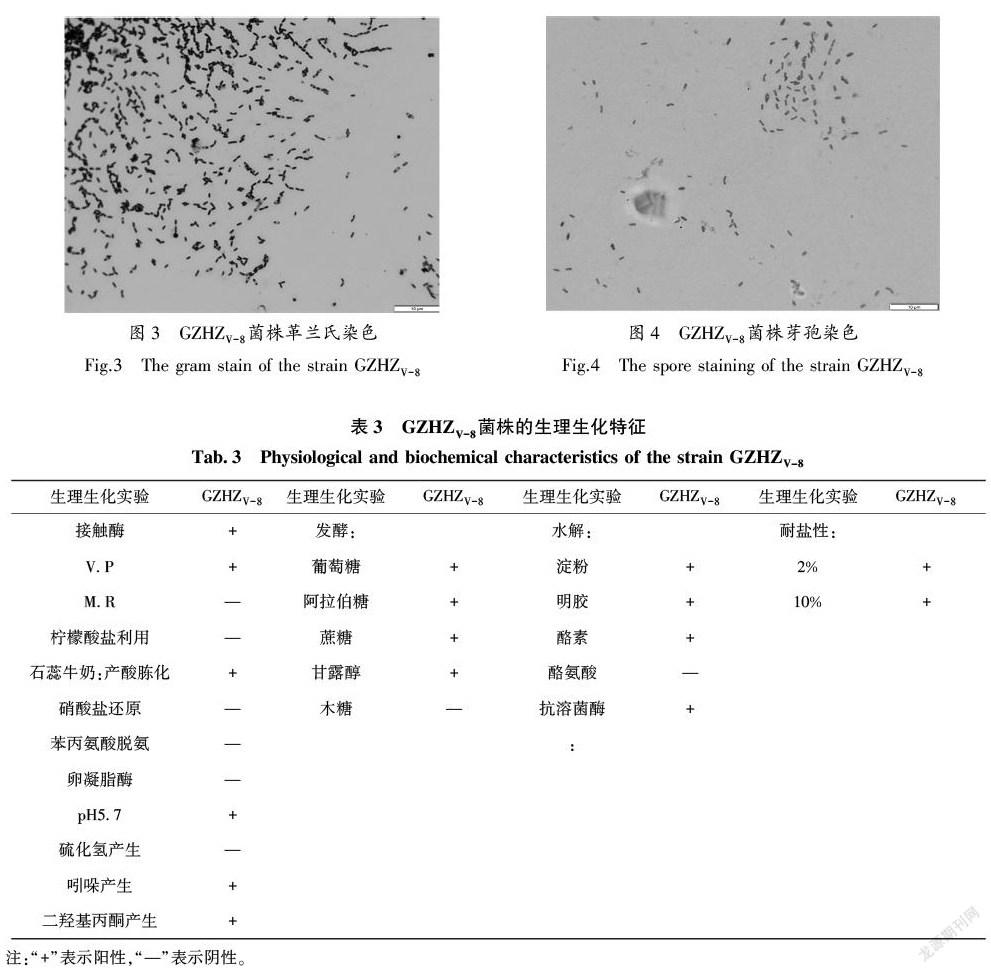
菌株GZHZV-8经镜检与菌落形态特征的观察,镜检为G+杆状细菌,两端椭圆,形成芽孢,芽孢中生或稍偏端生、柱状(图3),芽孢染色芽孢呈绿色,芽孢囊红色(图4)。单个菌落在牛肉膏蛋白胨培养基上培养24 h,菌落成圆形、乳白色、表面光滑湿润、有光泽、不易挑取。
2.4.2 生理生化鉴定
2.4.3 分子生物學鉴定
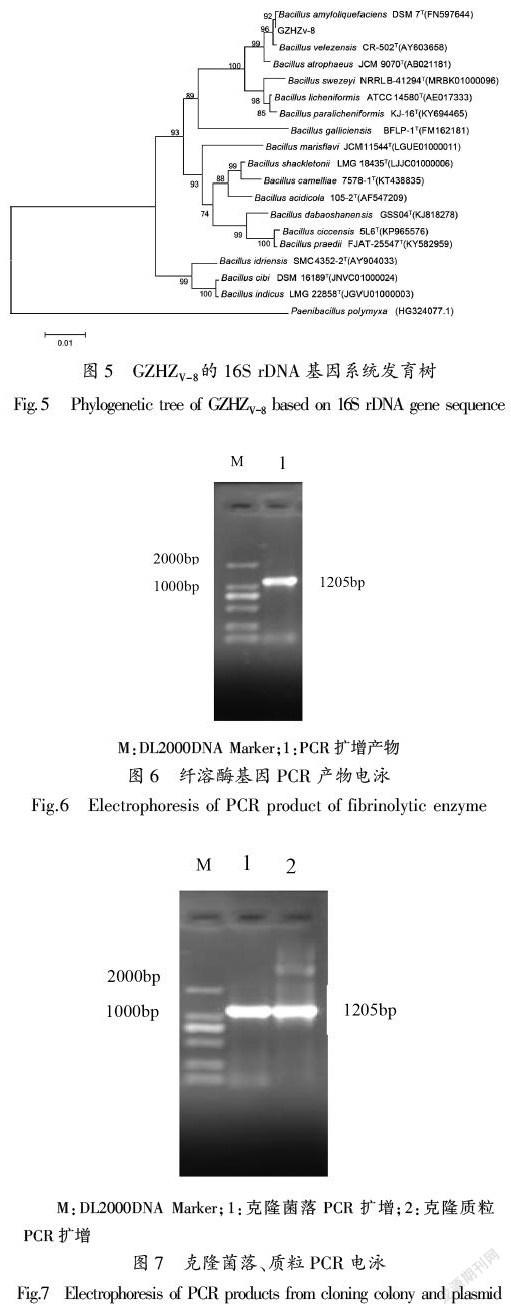
PCR产物经凝胶电泳检测,在1300bp处出现特异性条带。PCR扩增获得的16S rDNA基因序列与相关模式菌株构建系统发育树,株菌GZHZV-8的16S rDNA基因序列与其相似度较高的模式菌株聚成1个小分支(图5),GZHZV-8与解淀粉芽孢杆菌(Bacillus amyloliquefaciens)DSM 7(T)(FN597644)相似度为99.92%。结合形态学特征和生理生化特征以及分子生物学鉴定,该菌株可鉴定为解淀粉芽孢杆菌(Bacillus amyloliquefaciens)。
2.5 纤溶酶基因的扩增
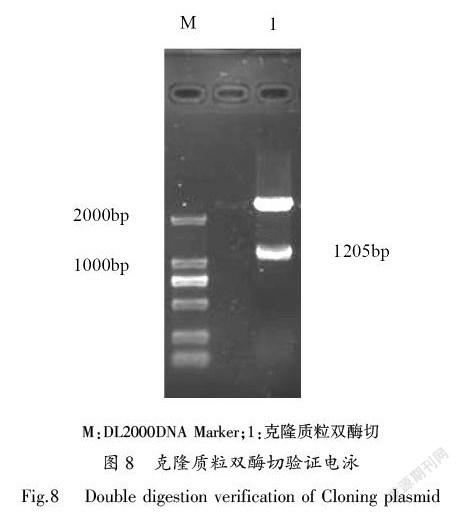
PCR扩增产物经1%的琼脂糖凝胶电泳检测,发现在约1200 bp左右有一条亮带(图6),与纤溶酶基因克隆设计引物的PCR产物大小一致,测序结果表明,克隆序列长度为1205 bp。
2.6 重组子鉴定
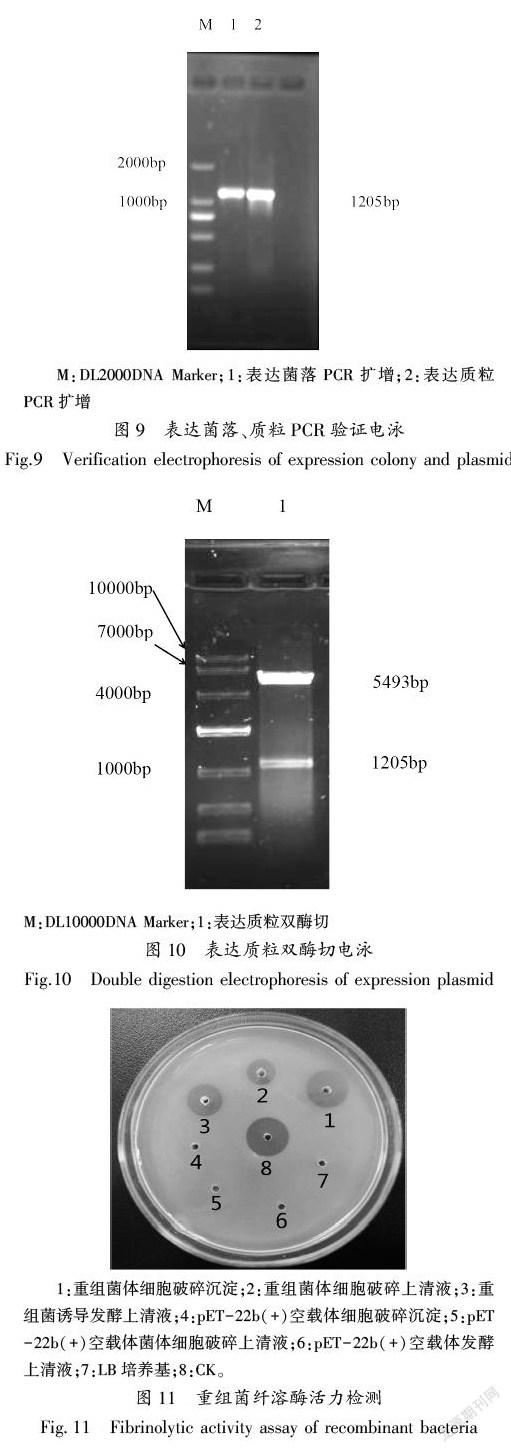
扩增纤溶酶基因与克隆载体连接后,经菌落PCR、质粒PCR、质粒双酶切验证,用1.0%凝胶电泳检测该片段大小为1200 bp左右,与预期片段大小相符。经测序验证纤溶酶基因已经准确的连接到克隆载体上(图7、8)。
2.7 纤溶酶基因序列与编码蛋白的理化性质的预测分析
利用NCBI的ORF Finder 在线分析,发现纤溶酶基因的编码区全长包含1092 bp开放阅读框,共编码363个氨基酸。将克隆的基因序列输入GenBank进行比对,结果表明,克隆的纤溶酶基因序列与已报道的纤溶酶基因序列核酸水平上相似度为97%,氨基酸水平上相似度为98%。突变了11个碱基,以致有3个氨基酸发生了改变。
利用ExPASy软件中ProtParam工具对纤溶酶基因编码蛋白的理化性质进行分析,蛋白理论分子式为C1621H2570N448O525S8,蛋白分子量为36.9914 kDa,理论等电点(PI)为8.73,含量最多的是丙氨酸(A)为13.8%,其次是丝氨酸(S)为12.7%,最低的是精氨酸(R)为0.6%。带负电荷的残基总数(Asp+Gly):25,带正电荷的残基总数(Arg+Lys):28。不稳定系数值26.29,是一种稳定蛋白。此蛋白的脂肪系数为78.51,亲水性的平均值为-0.105,表明该蛋白可能是亲水性蛋白。
2.8 重组表达质粒的构建与酶切
重组表达质粒经菌落PCR、质粒PCR、质粒双酶切经验证,1%凝胶电泳检测(图9、10),在1200 bp和5500 bp左右出现亮带,分别与pET-22b(+)和纤溶酶基因的大小相符,说明纤溶酶基因已正确插入表达载体。
2.9 重组纤溶酶的活力测定
经检测,重组表达质粒得到有活性的分泌性表达,根据尿激酶标准曲线计算出诱导发酵上清液酶活力为613.26 IU/mL,菌体破碎上清液的酶活力为407.38 IU/mL,菌体破碎沉淀酶活力为993.12 IU/mL(图11)。
3 结论与讨论
用大肠杆菌表达外源基因有许多优点,如大肠杆菌繁殖迅速,易于大规模发酵生产,表达外源基因的经验较为丰富[17]。有许多的研究者使用大肠杆菌表达系统来表达纤溶酶基因,如闰达中[18]将纳豆激酶基因克隆到载体pTYB102后,在大肠杆菌TR2566中表达出有活性的纳豆激酶,表达量达到30%以上。刘雪[19]将纤溶酶原基因连接到pET-27b(+)载体上,转化到大肠杆菌中,经葡萄糖诱导,能实现分泌活性的表达,发酵上清液纤溶酶活力为212.6 U/mL,活性蛋白分子量为28 kDa,但纤溶酶在大肠杆菌中的表达式为包涵体,分子量为38 kDa。因此,大肠杆菌表达系统对于纤溶酶的大规模生产具有重要意义。本研究以本实验室从酒曲、酒糟、窖泥等样品中分离的具有酱香味的13株菌株作为实验出发菌株,经脱脂奶粉平板、纤维蛋白双层平板筛选出纤溶酶活性最高的菌株GZHZV-8,结合形态学特征、生理生化特性实验及16S rDNA序列同源性分析,GZHZV-8菌株为解淀粉芽孢杆菌(Bacillus amyloliquefaci-ens)。用PCR技术扩增的GZHZV-8菌株纤溶酶基因与报道的纤溶酶基因在核酸与氨基酸水平上有高度的同源性,将纤溶酶基因与表达载体pET-22b(+)连接,构建重组表达载体,使纤溶酶基因成功转入大肠杆菌E.coli BL21,经IPTG诱导从而实现纤溶酶基因的表达。本研究结果表明,本实验室筛选的高活性菌株纤溶酶基因能在大肠杆菌中分泌表达。但重组菌纤溶酶活性与原始菌株相比,有所下降。pET-22b是利用T7启动子的高效原核表达载体。在根据Grossman[20]报道,葡萄糖能维持pET系统在λDE3宿主菌中低水平表达的蛋白,阻止T7溶菌酶的过量表达,且可以有效避免或延缓质粒丢失情况。在刘雪[19]的研究中验证了这一报道。罗文华等[21-22]分别利用枯草芽孢杆菌自生的启动子和枯草杆菌168的重叠启动子P43构建两种重组表达载体,分别在枯草芽孢杆菌WB800中表达,实验表明带P43启动子的重组菌纤溶酶活力是带枯草芽孢杆菌自身启动子的1.87倍。赵宇易等[23]报道了纤溶酶基因在大肠杆菌中的表达多以包涵体的形式存在。本研究中纤溶酶活力的降低,可能与启动子、表达宿主菌有一定的关系,刘雪与罗文华的报道[19,21-22]为本研究后续纤溶酶活性提高的研究提供了理论依据。
参考文献:
[1] 戴奕杰,李宗军,田志强.酱香型白酒大曲和糟醅的细菌多样性分析[J].食品科学,2018,1-16.
[2] 李 习,方尚玲,刘超等.酱香型白酒风味物质主体成分研究进展[J].酿酒,2012,39(3):19-22.
[3] 林 芬,谢 和.产酱香细菌中高活性纤溶酶菌株的筛选[J].食品科学,2010,31(17):258-262.
[4] Sumi H,Hamada H,Tsushima H. A Novel Fibrinolytic Enzyme (Nattokinase) in the Vegetable cheese Natto:A Typical and Popular Soybean Food in the Japanese Diet[J].Cellular and Molecular Life Sciences,1987,43(10):1110-1111.
[5] 傅 莉,李荣萍,李 晶.一株具有纤溶活性的枯草杆菌的研究液体发酵条件的选择[J].生物工程进展,1997,17(3):31-33.
[6] Nakamura T,Yamagata Y,Ichishima E. Nucleotide sequence of thesubtilisin NAT gene,aprN of Bacillus subtilis(natto)[J].Biosci Biotechem,1992,56(11):1869-1871.
[7] AI Hai-xin,ZHANG Li,ZHANG Xin-gang,et al. Prokaryotic expression,purification and characterization ofnattokinase[J].Journal of Microbiology,2017,37(1):20-27.
[8] 刘北域,宋后燕.纳豆激酶基因的克隆及其在枯草杆菌中的表达[J].生物化学与生物物理学报,2002(3):338-340.
[9] Lu Xiao,Ren-Huai Zhang,Yong Peng,et al. Highly efficient gene expression of a fibrinolytic enzyme (subtilisin DFE) inBacillus subtilis mediated by the promoter of α-amylase gene from Bacillus amyloliquefaciens[J].Biotechnology Letters,2004,26:1365-1369.
[10] 王开敏,赵 敏.产纤溶酶菌株的分离和鉴定及纤溶酶基因在酿酒酵母中的表达[J].中国食品学报,2009,9(2):24-28.
[11] 杨久玲,谢 和,杜超峰.一种酱香风味红茶的研制[J].山地农业生物学报,2017,36(6):90-94.
[12] Astrup T,Mullertz S. The fibri plate method for estimating fibrinolytic activity[J].Arch Biochem Biophys,1952,40:346-351.
[13] R.E.布坎南,N.E.吉本斯等.伯杰细菌鉴定手册[M].北京:科学出版社,1984.
[14] 东秀珠,蔡妙英.常见细菌系统鉴定手册[M].北京:科学出版社,2001.
[15] 王 欢,韩丽珍.4株茶树根际促生菌菌株的鉴定及促生作用[J].微生物学通报,2019,46(3):548-562.
[16] 佟硕秋,高 洁,钟 杰,等.烟草CYP71基因的克隆与原核优化表达[J].山地农业生物学报,2018,37(1):12-16.
[17] 柴玉波.大肠杆菌表达系统表达载体的类型及其研究进展[J].国外医学(分子生物学分册),1997,19(4):160-162.
[18] 闰达中,许 芳,李 洁,等.纳豆激酶基因克隆及其在大肠杆菌中活性表达研究[J].湖北大学学报:自然科学学版,2003(1):69-72,92.
[19] 刘 雪.豆豉纤溶酶产生菌的筛选及基因的克隆与表达研究[D].济南:山东师范大学,2011.
[20] Trudy H Grossman,Ernest S Kawasaki,Sandhya R Punreddy, et al. Spontaneous cAMP-dependent derepression of gene expression in stationary phase plays a role in recombinant expression instability[J].Gene,1998,209(1-2):95-103.
[21] 罗文华,郭 勇,韩双艳.豆豉纤溶酶在枯草杆菌WB800中的高水平表达[J].应用与环境生物学报,2007,35(11):565-569.
[22] 罗文华,郭 勇,韩双艳.枯草杆菌纤溶酶基因的克隆及表达[J].华南理工大学学报:自然科学版,2007,35(11):115-118.
[23] 赵宇易,刘晓兰,邓永平.微生物源纤溶酶基因的克隆及表达研究[J].齐齐哈尔大學学报:自然科学版,2016,32(6):68-71.
