Integrative analysis of the inverse expression patterns in pancreas development and cancer progression
2019-09-05HongLiangZangGuoMinHuangHaiYingJuXiaoFengTian
Hong-Liang Zang, Guo-Min Huang, Hai-Ying Ju, Xiao-Feng Tian
Abstract BACKGROUND As the malignant tumor, pancreatic cancer with a meager 5-years survival rate has been widely concerning. However, the molecular mechanisms that result in malignant transformation of pancreatic cells remain elusive.AIM To investigate the gene expression profiles in normal or malignant transformed pancreas development.METHODS MaSigPro and ANOVA were performed on two pancreas development datasets downloaded from the Gene Expression Omnibus database. Six pancreatic cancer datasets collected from TCGA database were used to establish differentially expressed genes related to pancreas development and pancreatic cancer.Moreover, gene clusters with highly similar interpretation patterns between pancreas development and pancreatic cancer progression were established by self-organizing map and singular value decomposition. Additionally, the hypergeometric test was performed to compare the corresponding interpretation patterns. Abnormal regions of metabolic pathway were analyzed using the Subpathway-GM method.RESULTS This study established the continuously upregulated and downregulated genes at different stages in pancreas development and progression of pancreatic cancer.Through analysis of the differentially expressed genes, we established the inverse and consistent direction development-cancer pattern associations. Based on the application of the Subpathway-GM analysis, we established 17 significant metabolic sub-pathways that were closely associated with pancreatic cancer. Of note, the most significant metabolites sub-pathway was related to glycerophospholipid metabolism.CONCLUSION The inverse and consistent direction development-cancer pattern associations were established. There was a significant correlation in the inverse patterns, but not consistent direction patterns.
Key words: Pancreatic cancer; Pancreas development; Inverse pattern; Metabolites subpathway
INTRODUCTION
As one of the most malignant tumors, pancreatic cancer results in more than 0.4 million deaths per year[1]. Generally, it is used to refer to pancreatic adenocarcinoma,which is the most common and devastating in all types of pancreatic cancer.According to the Union for International Cancer Control classification criteria,patients with pancreatic cancer can be divided into four main stages, including stage I, II, III and IV[2]. The overall five-year survival rate of the pancreas is less than 5% and the median survival period is six months after diagnosis, which is the lowest survival rate among all types of cancers[3,4]. Early diagnosis of biomarkers and effective treatments can help us effectively conduct pancreatic cancer research.
Similar to the tumor progression, organ development can also be divided into some stages in a time-dependent manner[5]. Moreover, the development is strictly controlled by multiple signaling pathways and transcription factors[6]. Some researchers believe that cancer is the problem of developmental biology. It has been reported that serval genes and pathways affected the normal or malignant transformed pancreas development[7]. Pancreatic and duodenal homeobox 1 (PDX1), which is exclusively expressed in the pancreas, is essential for pancreas development. Recently research shows that dysfunction of PDX1 also promotes pancreatic cancer development and progression[8,9].
We therefore investigated the relationship between pancreas development and pancreatic cancer progression by analyzing two datasets related with pancreas development and six datasets related with pancreatic cancer. Through bioinformatics analysis, we established differentially gene expression profiles and multiple patterns that were consistent/inverse in the development-cancer patterns. Moreover, we found that there was significant negative correlation between inverse development and cancer. Of note, we identified 17 metabolic sub-pathways that were highly related with pancreatic cancer development.
MATERIALS AND METHODS
Pancreas development datasets
To further understand the pancreas development, we analyzed two databases(accession: GSE42094, GSE96697), which obtained from the Gene Expression Omnibus(GEO) database. 16 samples in 6 development stages data were obtained from GSE42094, including undifferentiated human embryonic stem cells, stage 1 (S1), S2,S3, S4, and S5. In the GSE96697 database, according to the glycoprotein 2 (GP2) and CDH1 interpretation, we classified the 7-weeks pancreas development data into three stages, including an early, middle and late stage. We established that high interpretation of GP2 was early stage, while low interpretation was the middle stage.Additionally, CDH1 low interpretation of CDH1 was established as a late stage.
Pancreatic cancer datasets
To further understand the development and progression of pancreatic cancer, we analyzed five pancreatic cancer datasets which obtained from TCGA(https://portal.gdc.cancer.gov/), ICGC (https://icgc.org/) and GEO database[10-12].All of these datasets contained five or six clinical stages and available clinical information. The accession numbers in GEO database were GSE62452, GSE79668, and GSE102238. Detail information of the datasets was listed in Table 1.
Metabolite highly related to PACA
According to the previous studies, we established the differentially abundant metabolites and converted them to KEGG compound IDs[13-16]. These metabolites were obtained from the differentially abundant between control and pancreatic cancer. A total of 60 pancreatic cancer-associated differentially abundant metabolites were established.
Identification of differentially expressed genes in time-course datasets
We found the gene expression profile with significant differences between experimental groups in time course datasets using maSigPro approach. MaSigPro were applied to establish experimental groups using the dummy variables with a two-regression step, and it is an R package for significance analysis time-course microarray experiments. Briefly, after adjust, a global-scale regression model with all the defined variables, differently expressed genes was established. We applied a variable selection strategy to investigate the virtual difference between groups to establish the different profiles. According to the patient clinical stages, we could establish the pancreatic cancer interpretation profiles as “time-series” datasets. We selected 0.05 as false discovery rate (FDR) significance threshold for the analysis in this study without a special request. In GSE96697 dataset, due to the fewer development stage, we used ANOVA method to get differentially expressed genes(DEGs).
Identification of gene cluster with consistent expression patterns
To the investigation of the pancreas development and pancreatic cancer development and progression associated interpretation patterns, we performed an initial screening on the interpretation matrix of DEGs. We pre-processed the DEGs using a selforganizing map (SOM) strategy and performed the pattern recognition using the singular value decomposition (SVD) strategy. We conducted a comprehensive SOMSVD analysis under the guidance of as previous described (paper). Briefly, the analysis could conclude into three steps. Initially, we conducted the SOM transformation. Secondly, SOM output was decomposed using SVD strategy. FDR,which is used for significant neuron assessment, is set at 0.10 to select the pancreas development and pancreatic cancer progression pattern genes. The identified genes were analyzed using component plane presentation-integrated SOM for gene clustering. The highly similar expression pattern clusters were used for pancreas development and pancreatic cancer progression integrative analysis.
Function for Geneset enrichment analysis
In our study, we annotated the identified interpretation patterns using the Database for Annotation, Visualization and Integrated Discovery, which are comprehensive sets of functional annotation tools. The identified interpretation patterns were clustered into more than 40 Gene Ontology-Biological Process (GO-BP) terms. R package iSubpathwayMiner was applied for subpathway enrichment analysis.
Comparison of gene expression pattern between development and cancer
We identified the up-regulated interpretation patterns and down-regulated interpretation patterns in the pancreas development as dev-Up and dev-Dw. To analyze the associations between pancreas development and pancreatic progression,we use the hypergeometric test in our study, as previously described[17]. Briefly, we compared the dev-Up vs can-Up, dev-Up vs can-Dw, dev-Dw vs can-Up and dev-Dw vs can-Up to generated the development and cancer dataset pairs. Moreover, we considered that P < 0.05 was the significant correlation between pancreas development and pancreatic cancer progression. The p-value was calculated according to the following formula:
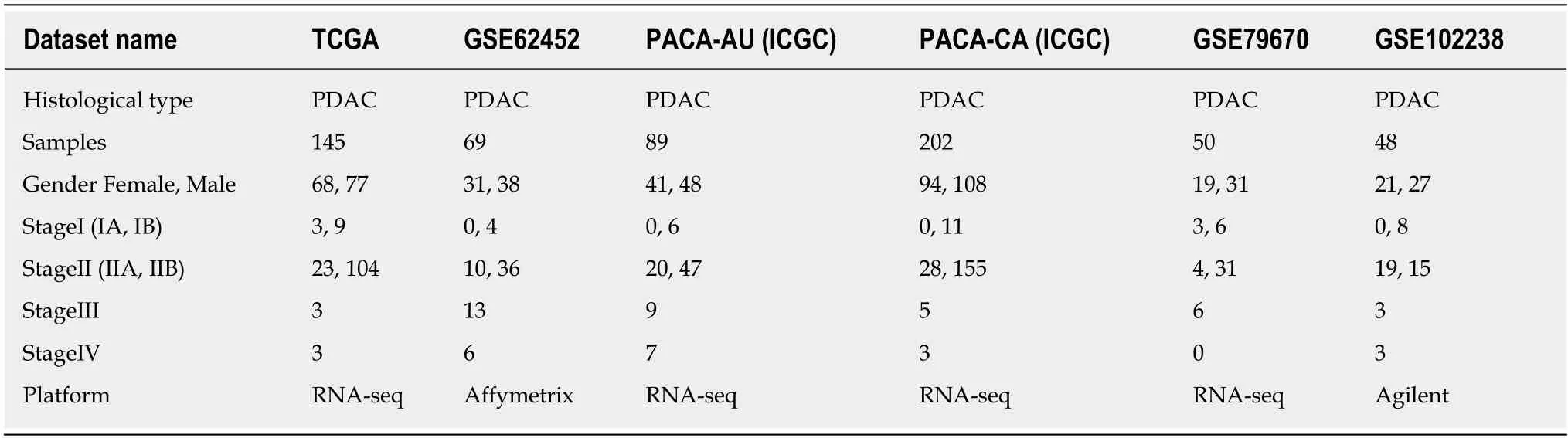
Table 1 Information of the pancreatic cancer datasets
In this formula, N-development and N-cancer indicate the number of pancreas development and pancreatic cancer regulation genes. M indicates the whole genome number, while k indicates the number of the common gene.
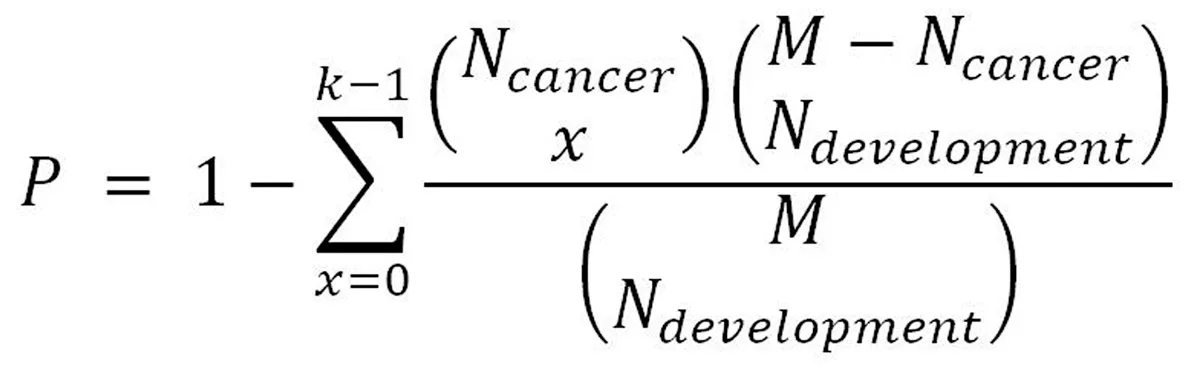
RESULTS
Identification of genes differentially expressed in pancreas development
To investigate the transcriptional pattern during pancreas development, we interrogated the interpretation patterns in GSE42094, GSE96697, which are the datasets of pancreas development. Using the maSigPro method, we identified 3069 DEGs at different time points of pancreas development in GSE42094 dataset. We then employed SOM-SVD strategy to select the topology-preserving DEGs, according to the interpretation matrices (Figure 1A). Reciprocally, the selected genes in a topologypreserving selection further confirmed the alteration in “time-series” processes. The entire genes were automatically selected in this method. Then, the matrices with 1257 genes, which obtained from SOM-SVD analysis, were clustered into four gene clusters(cluster 1-4). As shown in Figure 1A, the cluster 2 and cluster 4 contained genes that were transiently upregulated in the early stage of development, then decreased gradually along with development. We therefore named these gene sets as continuous down-regulated expression patterns. In contrast, genes with low expression level and were increased gradually in the latter stage of development, which were observed in cluster 1 and cluster 3. They were thus identified as continuously up-regulated expression patterns.
For the GSE96697 data set, we used the ANOVA method to analyze the expression data set and identified 3078 differential expressed genes. The K-means clustering method was used to establish patterns of interpretation of DEG sets, and we established six clusters. Similar to the previous continuous adjustment pattern recognition, we found that cluster 4 and cluster 6 are the interpretation modes of down-regulation, and cluster 2 is the interpretation mode of up-regulation (Figure 2).From this analysis, we found that 641 and 616 genes in the GSE42094 and GSE96697 data sets were continuously up-regulated, while 1059 and 1052 genes were continuously down-regulated. To investigate the biological characteristics of genes related with pancreatic development, functional enrichment analysis was used to aggregate genes in a consistent up- or down-regulation model. We found that ''lipid digestion' and ''cholesterol homeostasis'' were enriched during pancreas development and continued to up-regulate the mode of interpretation. "Cell proliferation," "mitotic nuclear division" is annotated in an interpretive pattern that is continuously downregulated (Figure 1B).
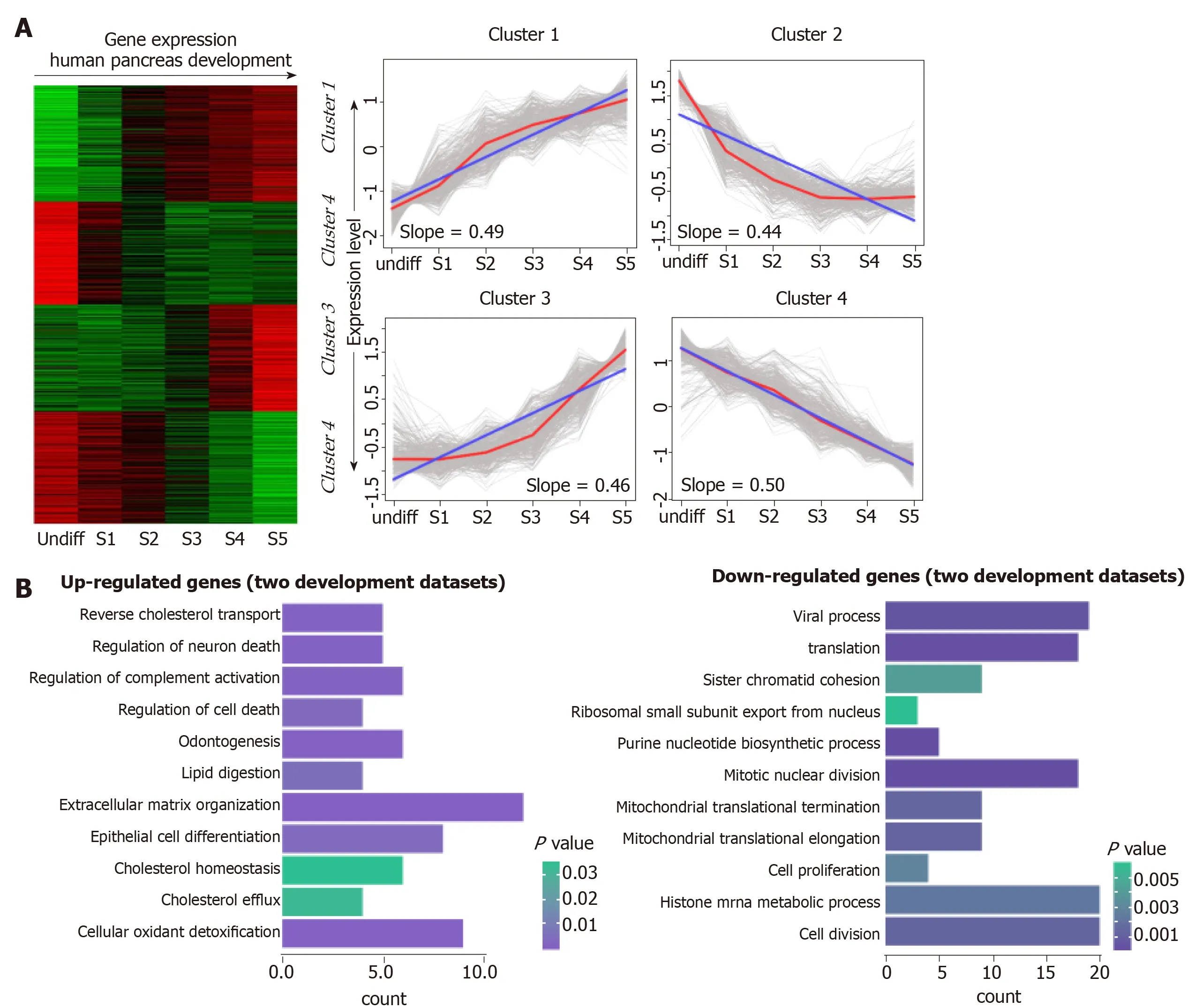
Figure 1 Continuous differential expression patterns in pancreas development. A: Clustering of human pancreas development genes. In the heat map, green indicates down-regulated, and red indicates up-regulated. In the line graphs, lines represent the tendency of the cluster changes; B: Gene Ontology-Biological Process annotations of the continuously up- and down-regulated genes.
Exploration of genes that continuously regulated in pancreatic cancer progression
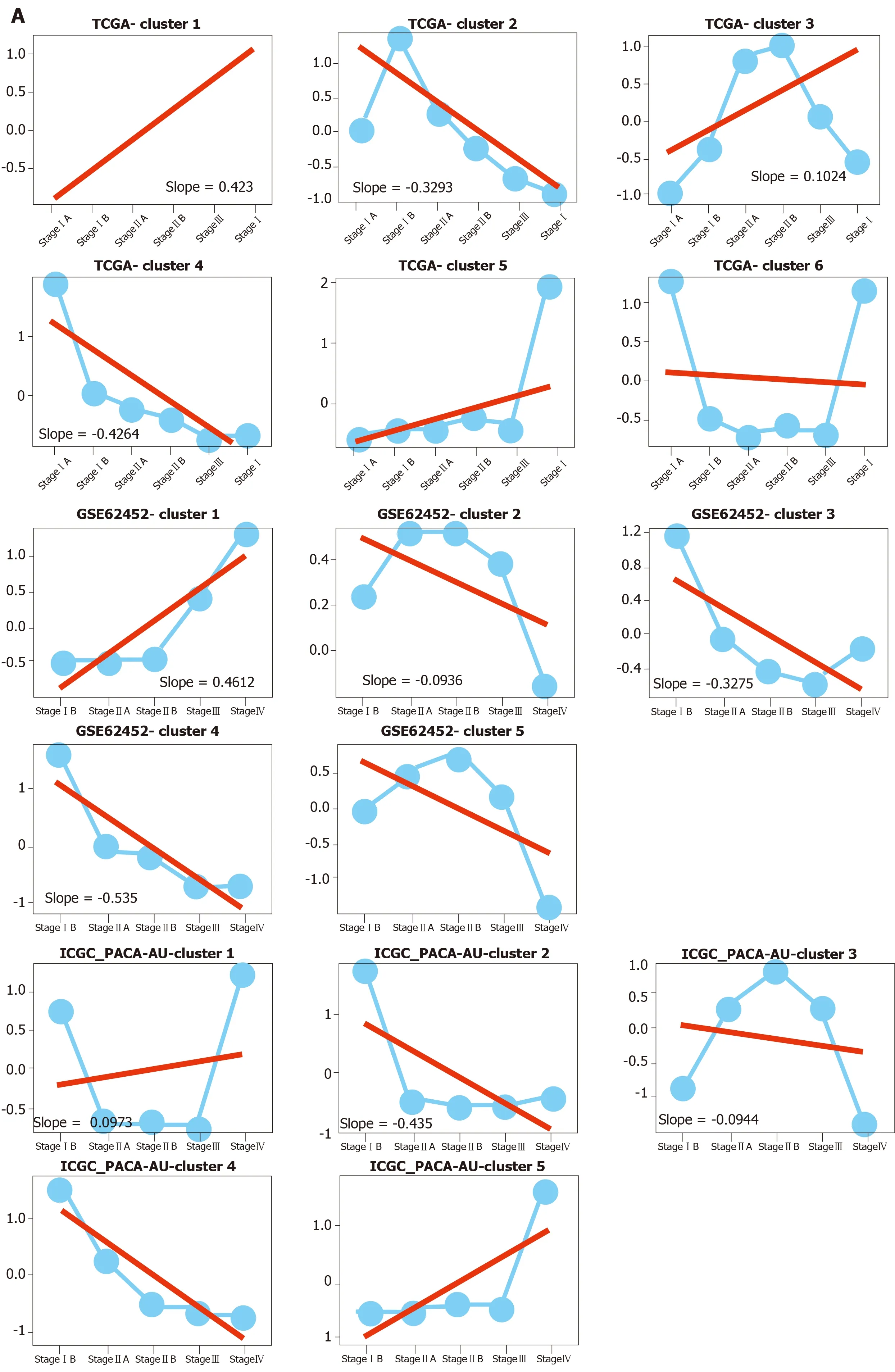
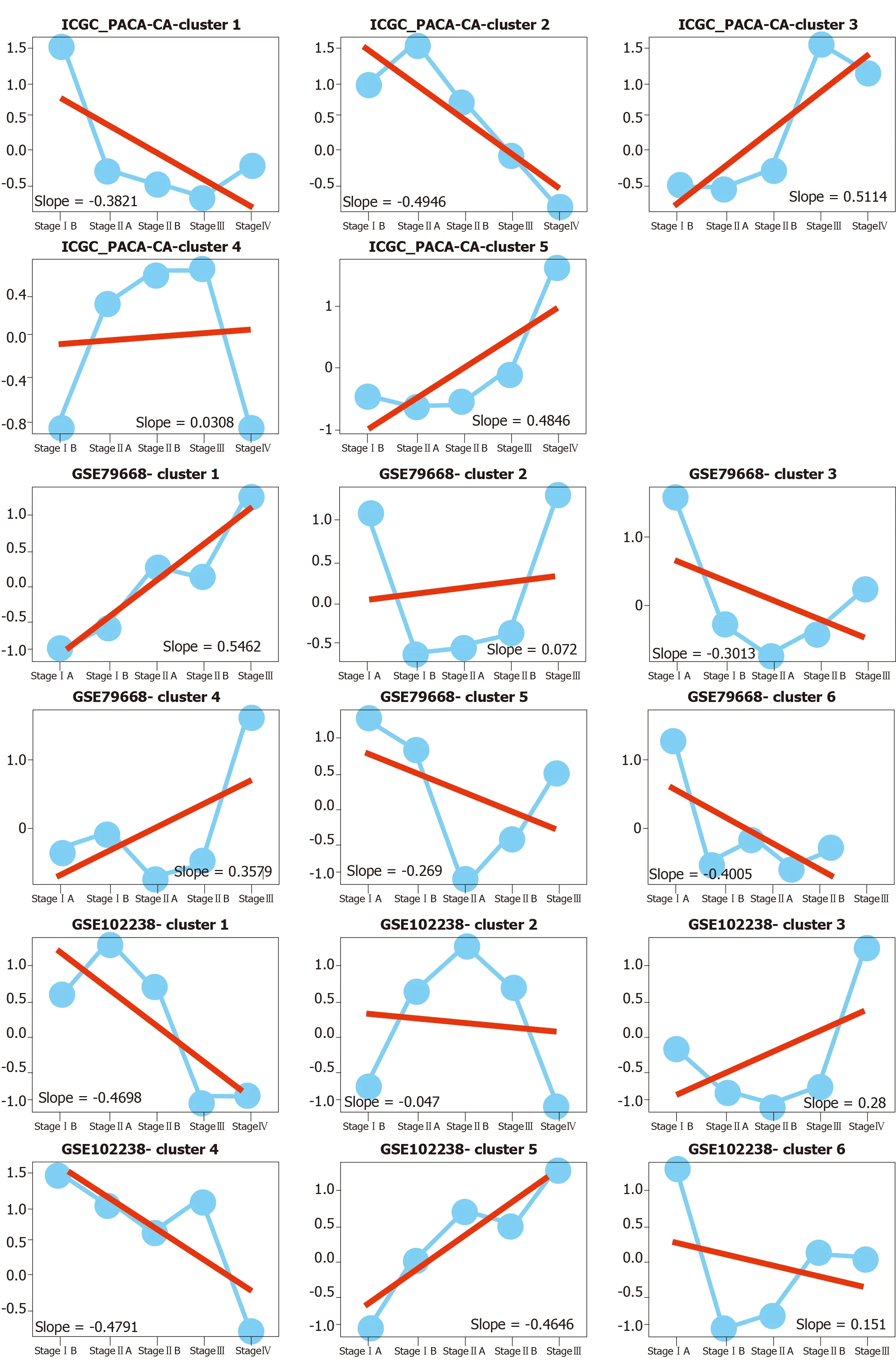
To measure the status of genes related with pancreas development in the progression of pancreatic cancer, we analyzed six pancreatic cancer data sets described in Materials and Methods. We considered each clinical stage of pancreatic cancer as the point in time to determine progression patterns, similar to pancreatic developmental analysis. Thus, we can determine the various interpretation patterns in these pancreatic cancer data sets (Figure 3A). Comparative analysis was used to study the mode of interpretation of continuous regulation between pancreatic development and pancreatic cancer progression. We established the interpretation mode of the upregulation of the tumor and the interpretation mode of the down-regulation according to the following criteria. Exceeding [(n-1)/ 2 + 1] the interpretation level of the adjacent stage changes with the same trend, n repeats the number of stages in each data set. The absolute slope is more significant than 0.05. We established six gene clusters in the up-regulated pattern and six of the down-regulated patterns (Figure 3B).
Comparative analysis of gene expression between pancreas development and pancreatic cancer progression
To investigate the relationship between pancreatic development and pancreatic cancer progression, we performed a hypergeometric test. As shown in Figure 4A, retropancreatic development and pancreatic cancer patterns, including dev-Up vs can-Dw and dev-Dw vs can-Up. On the other hand, we found a weak relationship between inconsistent development and cancer patterns. Specifically, we did not find any significant correlation between any two dev-Dw and can-Dw datasets. The inverse interpretation patterns were clustered into GO-BP terms to analyze the biological function, including dev-Up vs can-Dw and Dev-Dw vs can-Up patterns (Figure 4B).The results showed that 141 genes with dev-Up vs can-Dw were mainly associated with immune-related BP terms, including ‘‘T Cell Proliferation” and ‘‘Innate Immune Response In Mucosa''. Furthermore, 202 genes with dev-Dw vs can-Up were exclusively involved in proliferation-related BP terms, including “Cell Division” and‘‘DNA Replication Initiation''. Collectively, cell proliferation activity, as one of the essential characteristics in the malignant tumor, was gradually enhanced along with cancer progression, which was consistent with previous studies.

Figure 2 The expression patterns identified from GSE96697. Among these results, up- and downregulated patterns (red, green color labeled) were defined and further analyzed in our study.
Identification of Metabolic Sub-pathways Associated with pancreatic cancer
A total of 343 genes were established in the inverse interpretation patterns. To further establish pancreatic cancer associated with metabolic subpathways, we found 60 unique differentially abundant metabolites, which might be associated with pancreatic cancer progression. After integrating the analysis of 343 genes and 60 metabolites, Sub-pathway-GM strategy was employed to establish the critical,abnormal regions were identified in each metabolic pathway. Subsequently, we set FDR < 0.01 as a threshold for further analysis of 343 genes and 60 differential metabolite pathways and identified 17 significant metabolic sub-avenues (Table 2).Among these established sub-pathways, the most significant was"Glycerophospholipid metabolism" (path:00564_1) which was critical for lipid metabolism. Our data thus demonstrate that activation of metabolite pathways,especially lipid metabolism, are crucial for pancreatic cancer development.
DISCUSSION
Accumulative studies indicate that metabolism is substantial for cancer initiation and progression. Alterations of gene-related with metabolism in tumors provide increased energy for cancer cell proliferation even under nutritional deficient or hypoxia condition[18]. Integrative analysis of metabolic pathway and metabolites facilitates us to better understanding of the underlying mechanism and potential drug-targets of pancreatic cancer[13,19].
Following analysis of the pancreas development database and pancreatic cancer database, we established the 202 genes with dev-Dw and can-Up, which were mainly associated with cell proliferation. Consistent to previous studies, our results confirm that the uncontrolled proliferative activity of cancer cell is the most remarkable hallmarks of carcinogenesis, increased along with tumor progression[22]. Conversely,the proliferative capacity is decreased along with normal pancreas development[20-22].DNA replication is occurred accompanied by cell proliferation. Due to the infinite hyperplasia of cancer cell, the DNA replication is continuously up-regulated in pancreatic cancer[23,24]. Due to the limited proliferation of healthy organs, DNA replication continuously downregulates pancreatic development[25]. We established 141 genes with dev-Up, can-Dw, including ‘‘T Cell Proliferation” and ‘‘Innate Immune Response In Mucosa''. As a dynamic process, the carcinogenesis, associated with immunoediting[26]. Along with cancer progressed, the host immunosurveillance were suppressed, which in turn led to cancer immune escape. Interestingly, though analysis of metabolism pathways, we found that genes related with steroid hormone biosynthesis were dysregulated during cancer development. Steroid can elicits immunosuppressive effects and restricts T cell-mediated cancer eradication[27]. We therefore hypothesized that enhanced steroid production were the primarily cause for cancer immunoediting.
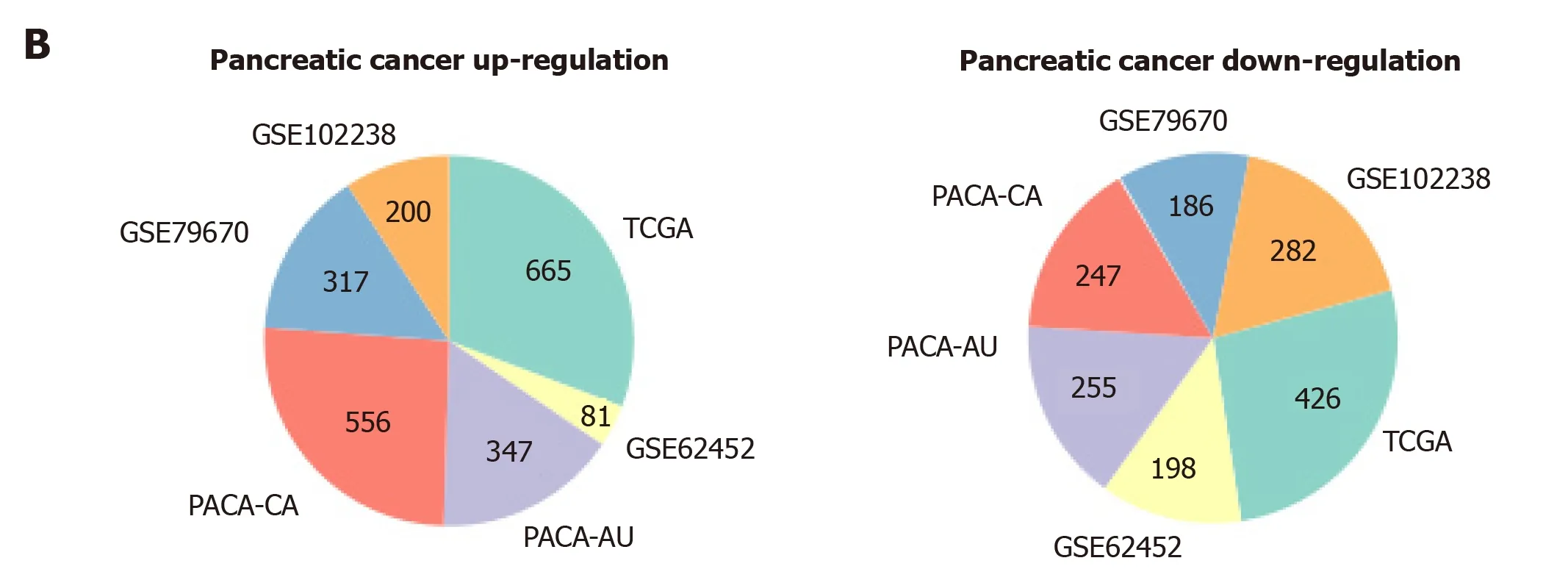
Figure 3 The expression patterns identified from six pancreatic cancer datasets. A: Among these results, up- and downregulated patterns (red, blue color labeled) were defined and further analyzed in our study; B: Pie diagram showed the up- and down-regulation genes in each pancreatic cancer dataset.
After analysis of the pancreas development database and pancreatic cancer database, we established that Glycerophospholipid metabolism, as an essential subpathway in lipid metabolism, was the most significant sub-pathway. Many pieces of research have been shown that lipid metabolism was strongly linked with pancreas cancer. The pancreatic lipase, as a lipolytic enzyme, was thought to be one of the predictors for prognosis and cancer-specific mortality in pancreas cancer[13]. Pancreatic lipase 1 and 2 is a significant lipase for lipid hydrolysis in pancreatic cancer patients,which is significantly reduced compared to healthy controls. Additionally, the inversed expressed genes and metabolites in the glycerophospholipid metabolism are associated with pancreatic cancer. SLC44A4, closely associated with acetylcholine synthesis and transport, was markedly upregulated in advanced and undifferentiated epithelial tumors, especially in prostate and pancreatic cancer[28,29]. Choline could decrease the risk of developing pancreatic cancer[29]. Besides, choline and phosphocholine have been reported to be associated with other cancers, including breast cancer, ovarian tumor[30-31]. Some researchers believe that increased choline and phosphocholine are the critical aspects of tumor metabolism and tumor cell migration[32]. Besides lipid metabolism, recent studies also reveal that activation of glycolysis and citrate cycle pathway promotes cancer development[33].
Above all, here we identified a series of gene related with metabolism via bioinformatics analysis, which are crucial for cancer development. We believe that our findings may provide potential targets for the treatment or prognosis of pancreatic cancer.
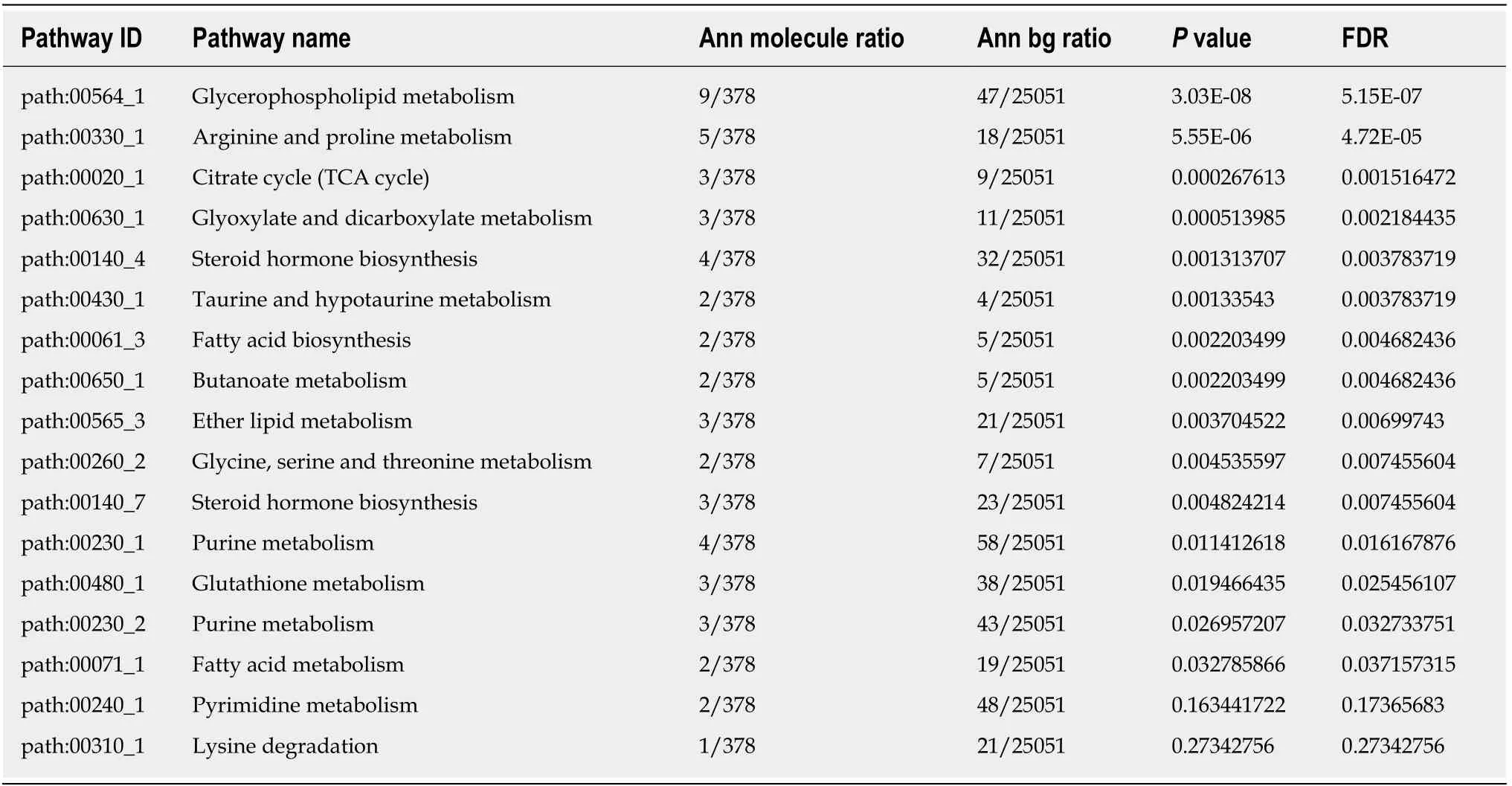
Table 2 17 significant metabolic subpathways in pancreatic cancer
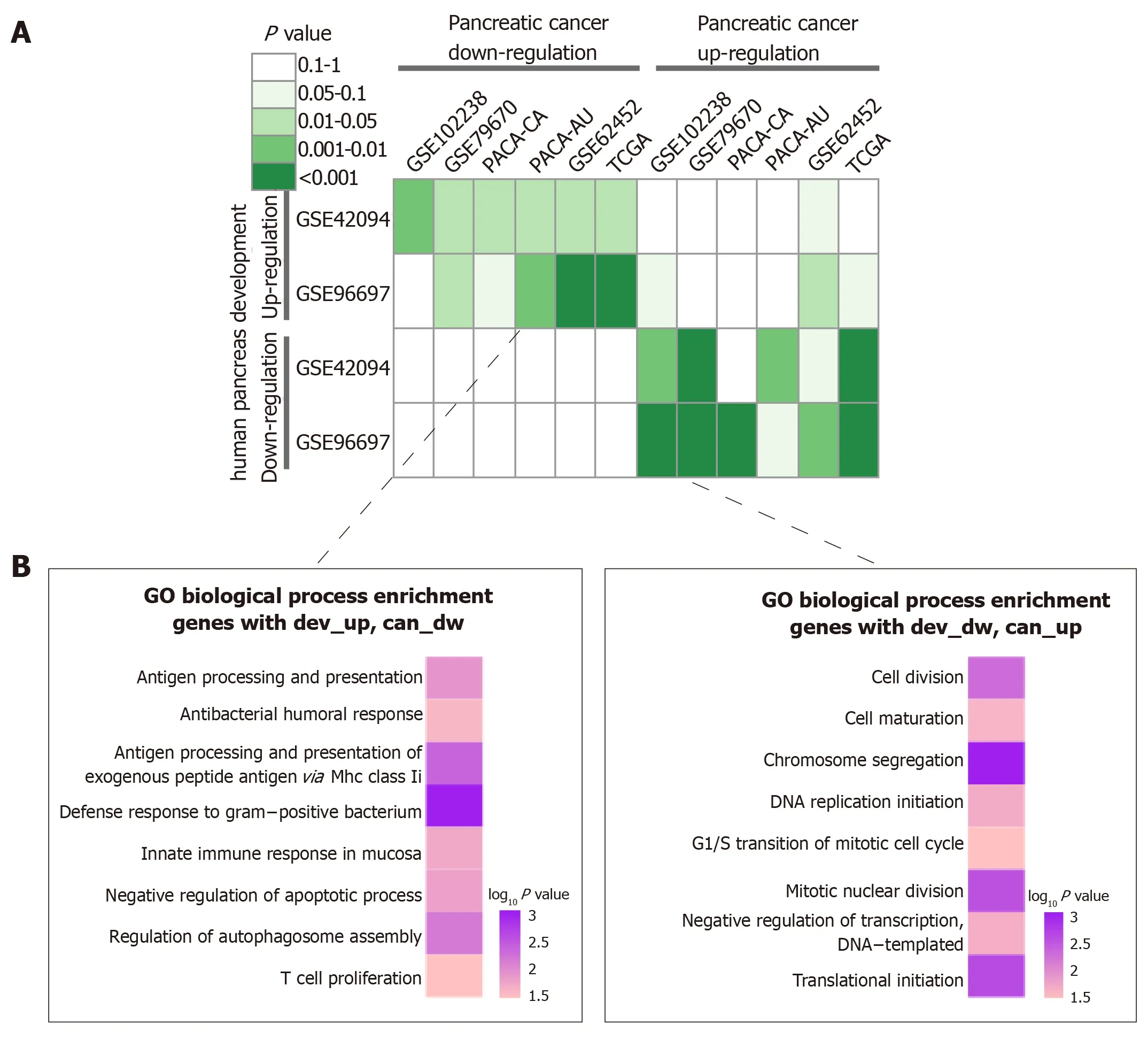
Figure 4 Integrated analysis of expression patterns in pancreas development and pancreatic cancer progression. A: Comparison of interpretation patterns between pancreas development and cancer progression. Color in each cell indicated the p-value; B: Gene Ontology-Biological Process annotations of the inverse interpretation patterns, including dev-up vs can-dw and dev-dw vs can-up.
ARTICLE HIGHLIGHTS
Research background
Pancreatic diseases remain as one of the most feared and clinically challenging diseases to treat despite continual improvements in therapies.
Research motivation
To develop agents into a targeted drug for explicitly killing cancer cells.
Research objectives
To explore the molecular interpretation patterns of pancreas development and cancer progression.
Research methods
This study used the ANOVA method, self-organizing map-singular value decomposition analysis, enrichment analysis, and hypergeometric test.
Research results
The results investigate continuously dysregulated interpretation patterns in pancreas development and pancreatic cancer.
Research conclusions
Integrative analysis of continuously dysregulated interpretation patterns to establish the inverse interpretation in metabolites and gene levels. Through integrating the genes with metabolites,some key abnormal regions of metabolic pathways have been established.
Research perspectives
With the increase of human disease database, a larger-scale integrative analysis is needed for the correlation with pancreas development and cancer. We believe the more convince underlying mechanism and potential drug development targets could be supposed by the larger-scale development and integrative cancer analysis in further. Also, this method could be used for other diseases investigation.
ACKNOWLEDGEMENTS
We appreciate Dr. Xi Huang for supports in editing the manuscript.
杂志排行

World Journal of Gastroenterology的其它文章
- New Era: Endoscopic treatment options in obesity-a paradigm shift
- Chronic hepatitis delta: A state-of-the-art review and new therapies
- Eosinophilic esophagitis: Current concepts in diagnosis and treatment
- Locoregional treatments for hepatocellular carcinoma: Current evidence and future directions
- Review of current diagnostic methods and advances in Helicobacter pylori diagnostics in the era of next generation sequencing
- Exploring the hepatitis C virus genome using single molecule realtime sequencing