Chronic hepatitis delta: A state-of-the-art review and new therapies
2019-09-05ChristyGilmanTheoHellerChristopherKoh
Christy Gilman, Theo Heller, Christopher Koh
Abstract Chronic delta hepatitis is the most severe form of viral hepatitis affecting nearly 65 million people worldwide. Individuals with this devastating illness are at higher risk for developing cirrhosis and hepatocellular carcinoma. Delta virus is a defective RNA virus that requires hepatitis B surface antigen for propagation in humans. Infection can occur in the form of a co-infection with hepatitis B, which can be self-limiting, vs superinfection in a patient with established hepatitis B infection, which often leads to chronicity in majority of cases. Current noninvasive tools to assess for advanced liver disease have limited utility in delta hepatitis. Guidelines recommend treatment with pegylated interferon, but this is limited to patients with compensated disease and is efficacious in about 30% of those treated. Due to limited treatment options, novel agents are being investigated and include entry, assembly and export inhibitors of viral particles in addition to stimulators of the host immune response. Future clinical trials should take into consideration the interaction of hepatitis B and hepatitis D as suppression of one virus can lead to the activation of the other. Also, surrogate markers of treatment efficacy have been proposed.
Key words: Hepatitis delta; Epidemiology; Treatment
INTRODUCTION
Hepatitis D virus (HDV) infection was initially identified in 1977 in Italy by Dr. Mario Rizzetto and colleagues who noted a severe hepatitis in individuals thought to be mono-infected with hepatitis B virus (HBV). Initial immunofluorescence studies led to the finding of a novel antigen which was thought to be a new biomarker for HBV[1].Serum analysis of these affected patients showed that they developed an antibody to a newly identified antigen which was subsequently termed the delta antigen (HDAg).In the early 1980s, collaborative studies in chimpanzees revealed more information regarding this novel finding. Hepatitis B susceptible and immune animals were inoculated with hepatitis B surface antigen (HBsAg) positive sera from patients with HDAg and only susceptible animals developed HBV and delta markers. Also,synthesis of delta markers caused the synthesis of HBV gene products to be diminished. This study importantly highlighted that HDV was transmissible, requires HBV for its own replicative cycle and interferes with HBV replication[2]. Over the past 40+ years since the initial identification of HDV, chronic HDV infection has come to be known as the most severe form of viral hepatitis[3]which leads to higher rates of cirrhosis[4]and hepatocellular carcinoma (HCC)[5]compared to HBV mono-infection[6-8]and chronic hepatitis C (HCV) infection[9]. To date, there is no treatment approved by the United States Food and Drug Administration (FDA) for chronic HDV infection.Current treatment with interferon alpha, as recommended by professional societies,has limited efficacy thereby leaving liver transplant a final viable option. However,new and promising treatment options are being investigated in clinical trials.
EPIDEMIOLOGY
Approximately 587 million people are infected with chronic HBV infection with 62-72 million chronically infected with HDV[10]. Eight HDV genotypes have been identified to date with genotype 1 being widely distributed, 2 and 4 predominantly in Asia, 3 localized to the Amazon Basin and 5-8 of African lineage (Figure 1)[11,12]. Prevalence rates vary widely with high rates noted in Africa and South America (Table 1).Retrospective studies have noted that HDV infection is more common in men than women[13,14]. High risk populations include individuals from endemic areas,intravenous drug users, men who have sex with men, individuals with human immunodeficiency virus (HIV) or HCV and patients with multiple sexual partners[5,13,15,16]. In a retrospective analysis in Spain, there was a reduction of new cases of acute HBV and HDV infection between the time periods 1983-1995 and 1996-2008 and it is thought that policy changes during the 1990s for HBV vaccination to include all newborns and adolescents and not just high risk groups played a substantial role[13,17]. Immigration and sexual transmission are now the leading risk factors for HDV infection[17].
In the United States, testing for HDV infection has been poor[4,15,18]. In a retrospective study of 1191 patients in northern California from 1989-2007, only 499 (42%) patients were tested for HDV (HDV ab or HDAg or both)[4]. Of those tested, 42 (8%) patients were confirmed to be HDV infected [HDV ab (n = 29), HDAg (n = 6), both (n = 7)].Interestingly, 67% of the HDV patients were diagnosed with cirrhosis compared to only 17% of HBV monoinfected patients tested for HDV and 22% of the total HBV monoinfected cohort (including patients not tested for HDV). In a retrospective study of patients in the Veterans Affairs medical system from 1999-2013, 2008 (7.8%) of 25,603 HBsAg positive patients were tested for HDV and 73 (3.6%) had a positive HDV ab[5]. In a cross-sectional study of electronic medical records from 1994-2014, 121(12%) of 1007 HBsAg positive were tested for HDV and 4 (3.3%) had a positive HDV ab[18]. These studies highlight the need for HDV screening in all patients with HBV in congruence with the Asian Pacific Association for the Study of the Liver (APASL)[19]and European Association for the Study of the Liver (EASL)[20]guidelines for education, possible need for treatment and prevention of transmission.

Table 1 Prevalence rates throughout the world
VIROLOGY
HDV is the smallest known human RNA virus and is a defective RNA virus which requires HBsAg[21]. It is about 36 nm in diameter and consists of a circular single stranded RNA (about 1700 BP)[22], that folds into a rod like structure[23]due to selfcomplementarity[24], and HDAg thus forming the HDV ribonucleoprotein (RNP)[25]surrounded by the HBsAg envelope (Figure 2)[21,26,27]. Entry of the HDV RNP into hepatocytes occurs through binding of the sodium taurocholate co-transporting polypeptide (NTCP) receptor[28,29]through the preS1 region of the large HBsAg. Once in the hepatocyte, transport to the nucleus is mediated by HDAg[25,30]through a nuclear localization signal[31-34]and possibly through phosphorylation[35], acetylation[36]and methylation[37]of HDAg. Replication occurs through the host RNA polymerase[38-41]in a double-rolling cycle[22]. Rolling cycle replication allows for transcription of full-length antigenomic RNA which is used to generate genomic RNA. The antigenomic RNA contains the sequence for HDAg[37,42], which undergoes RNA editing and self-cleavage[43,44], and translation occurs in the endoplasmic reticulum. HDAg is present in two forms based on RNA editing[44]and are referred to as small (SHDAg, 195 amino acids, 24 kDa) and large (LHDAg, 214 amino acids, 27 kDa) delta antigen[42]. This editing process adds additional amino acids to the Cterminus of LHDAg[45]. Replication is promoted by SHDAg[46,47]. LHDAg suppresses SHDAg[47], contains an isoprenylation motif and nuclear export signal[48,49]and promotes assembly of the virus[46,50-52]. Genomic RNA is exported to the cytoplasm through signaling in HDV RNA[34]. LHDAg promotes prenylation and association with HBsAg[53]generating a viral particle. SHDAg alone is insufficient for virion formation and it is thought that the LHDAg acts as a bridge between HBsAg and SHDAg and HDV RNA[25,34,51,52,54,55](Figure 3).
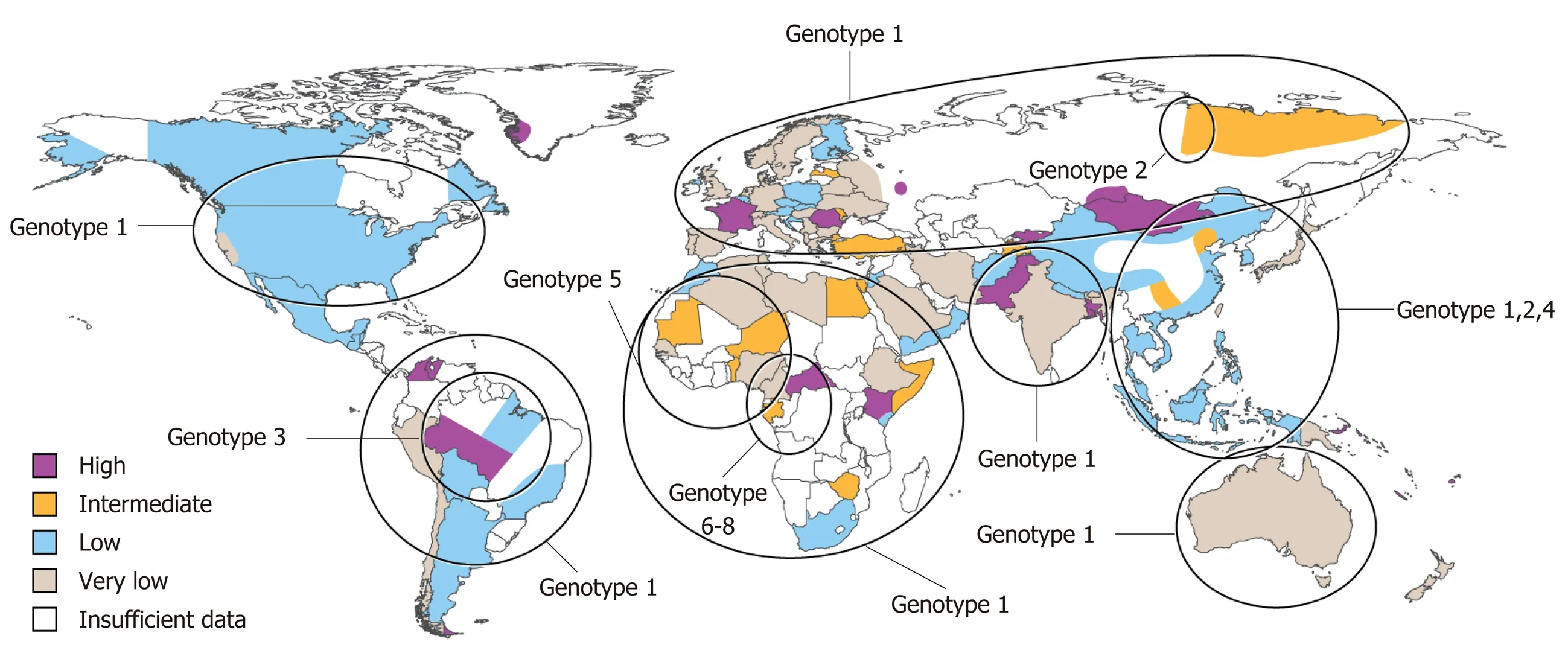
Figure 1 World map illustrating the estimated prevalence of hepatitis D virus infection with genotypic distribution.
PATHOGENESIS
Studies have shown that there is an interaction between HBV and HDV though the exact mechanism has not been elucidated. In a longitudinal analysis of 33 chronic HDV patients, HDV was the predominant replicating virus in 54.5% of cases, whereas HBV was the predominant replicative virus in 30.3% of cases and both were codominant 15.2% of cases[56]. Compared to HBV mono-infection, it has been reported that HBV/HDV infection leads to more severe liver damage[8,57-59]including extensive necrosis[3,60-62]and patients develop cirrhosis at a faster rate[8]. Also, though suppressed,low level HBV replication may be capable of causing liver damage[63]. In chimpanzees,damage by HBV is immune mediated whereas in HDV it is cytopathic[64]or possibly cytotoxic as evidenced with a predominance of macrophages in areas of necrosis[62].An in vitro model to determine the effect of HDAg expression in HeLa and HepG2 cells showed that HDAg is directly cytotoxic[65]. In a study analyzing cytokine responses in patients with chronic HDV infection before and during interferon alpha(IFN-α) treatment, it was found that interleukin (IL)-2, interferon gamma (IFN-γ),interferon-inducible protein-10 and IL-10 responses were detectable and that these responses declined during treatment with interferon[66]. In a humanized mouse model evaluating the antiviral state of human hepatocytes in the setting of HBV/HDV coinfection compared to HBV mono-infection, the JAK STAT pathway was triggered by HDV infection[67]. Natural killer cells were studied in chronic untreated HDV patients[68]. Elevated STAT1 levels were observed in chronic HDV patients and treatment with pegylated interferon alpha (peg-IFN-α) further augmented this elevation and reduced induction of STAT4 signaling with expected decrease in IFN-γ.An integrated genomic approach of liver specimens from HDV-HCC (n = 5) and non-HCC HDV cirrhosis (n = 7) at the time of liver transplantation showed that HDV-HCC tissue exhibited upregulated gene expression of cell cycle pathways important for DNA replication, damage and repair and the pattern is disease specific[69]. A more recent study suggests that the pathways to damage of hepatocytes may be due to the imbalance in type 1 and 2 immunity, activated HDV-specific CD8+T cells and presence of escape variants[70,71].
CLINICAL PRESENTATION AND DIAGNOSIS
It is important to have a high clinical suspicion for HDV especially in high risk patient populations and HBsAg positive individuals with high alanine aminotransferase(ALT) and low or undetectable HBV DNA levels[5,63,72]. The recommended screening test is anti-HDV antibodies with follow up HDV RNA testing if screening is positive[15]. The clinical presentation of HDV can be variable and patients can be asymptomatic with mild enzyme elevation vs fulminant hepatic failure. HDV can present as a co-infection vs superinfection with HBV. The main serological difference between them is positive anti-HBc IgM in co-infected individuals[64,73]. Co-infection occurs with acute infection of both viruses. A biphasic pattern of ALT and bilirubin can be observed[74]. Clinical course is typically benign, similar to acute HBV infection,spontaneous recovery occurs in up to 90% of individuals and progression to chronicity occurs in 2-8% of individuals[13,73,75,76]. Superinfection occurs in individuals with chronic HBV infection who are later infected with HDV. Spontaneous recovery occurs in 10% of individuals with progression to chronicity in 90%-100% of individuals[13,73,75]. Chronic HDV infection leads to cirrhosis in about 80% of individuals in 5-10 years[4,77].
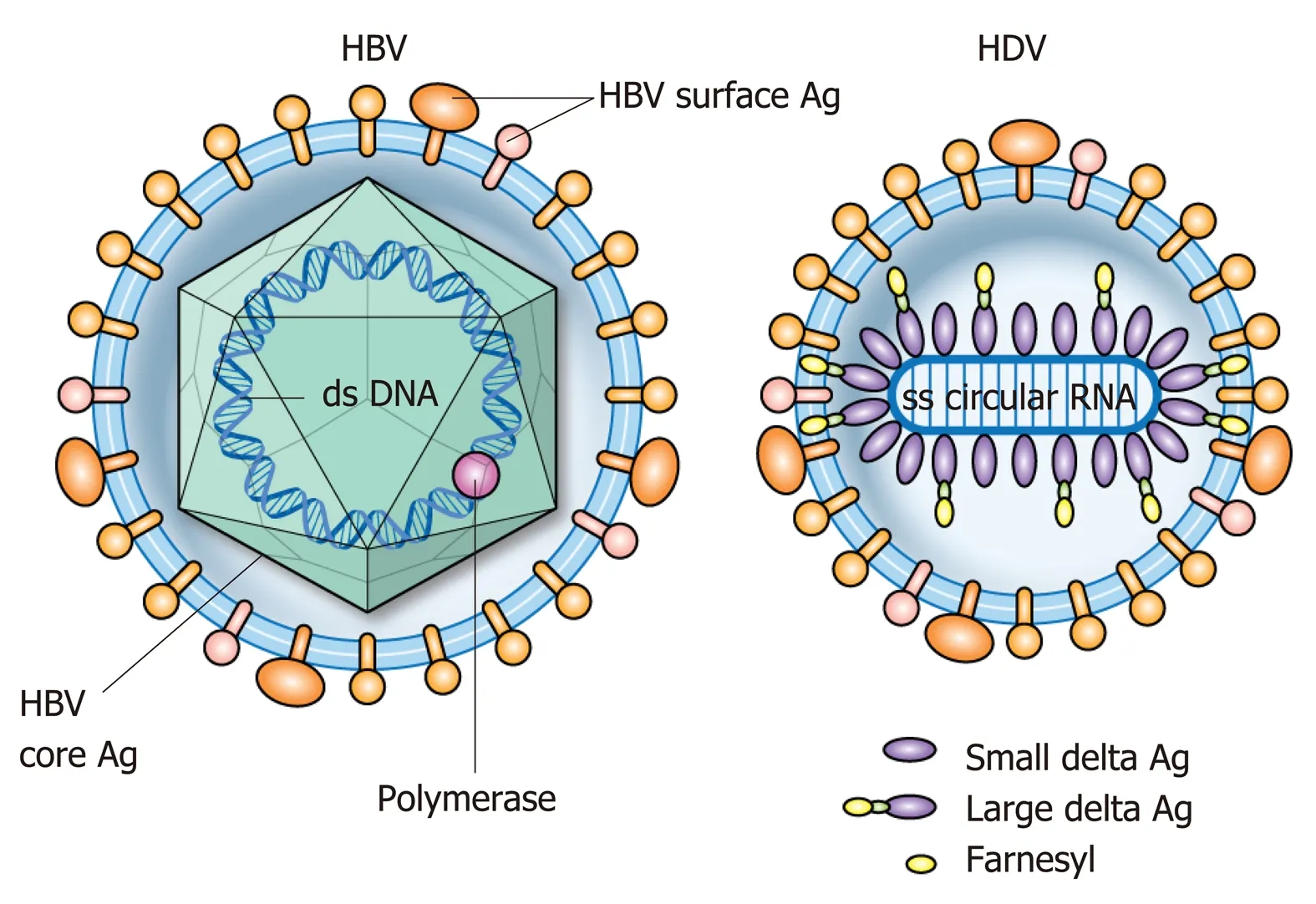
Figure 2 Structural representation of hepatitis B and delta viruses.
Disease progression is striking in patients with chronic HDV infection[57]and scoring systems in addition to noninvasive markers of fibrosis have been evaluated for predicting damage and clinical outcomes. The baseline-event-anticipation score(BEA score) resulted from a 16-year study of 75 HBsAg-anti-HDV-positive patients with delta hepatitis and validated in two independent cohorts[78]. Age, sex, region of origin, bilirubin, platelets and international normalized ratio (INR) and based were included and allocated points and the summation resulted in mild, moderate and high risk groups[78].
The Delta Fibrosis Score (DFS) was developed in a cohort of 100 HDV RNA positive patients in Europe[79]. Eight non-invasive fibrosis scores were evaluated (aspartate aminotransferase (AST) to ALT ratio (AAR), AST to platelet ratio index (APRI),fibrosis-4 score (FIB-4), Non-alcoholic fatty liver disease (NAFLD) score, BARD score,European Liver Fibrosis (ELF) score, SHASTA index, Amino-terminal propeptide of type III collagen) and compared to liver biopsy using the Ishak scoring system. F3-F4 indicated advanced fibrosis and F5-F6 indicated cirrhosis. Goal AUROC was > 0.8,sensitivity > 80% and positive predictive value (PPV) > 90%. None of the scores met criteria even after adapting cut-off values to optimize for chronic HDV infection. The ELF score did meet criteria of sensitivity and PPV but was determined to not be cost effective especially for endemic regions with limited sources of funding. Thus, the DFS was developed and components included older age, elevated gamma glutamyl transferase (GGT), low albumin and low cholinesterase with an AUROC of 0.87,sensitivity of 85% and PPV of 93%.
The utility of serum fibrosis markers in HDV were also explored in a retrospective,cross-sectional study of 1003 patients with chronic hepatitis B (n = 240), C (n = 701)and D (n = 62) who underwent liver biopsy[61]. Markers included FIB-4, AAR, ageplatelet index (API), APRI and Hui score. Advanced fibrosis was defined as Ishak ≥ 4 and cirrhosis as Ishak 6. FIB-4 appeared to perform the best in this comparison with an AUROC of 0.70, although the performance was substantially better in the comparative HBV and HCV cohorts. Current therapy for chronic HDV infection is limited to interferon alpha. Identifying patients who warrant treatment highlights the need for specific HDV scoring systems and fibrosis markers.
MANAGEMENT
Currently there is no treatment approved by the United States FDA for chronic HDV infection. Current guidelines of the American Association for the Study of Liver Diseases (AASLD), Asian Pacific Association for the Study of the Liver (APASL) and the European Association for the Study of the Liver (EASL) recommend peg-IFN-α for 12 mo[15,19,20]. IFN-α, peg-IFN-α and combination therapy with nucleos(t)ide analogues(NA) have been studied in chronic HDV infection but therapy with IFN is largely limited to patients who are non-cirrhotic. Novel treatment options have been evaluated recently and four areas of interest include drugs that target entry, assembly and release of viral particles in addition to treatment that activates the immune host response.
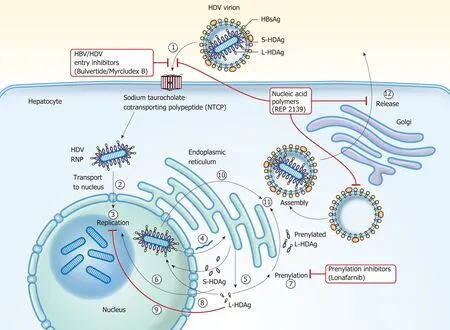
Figure 3 Hepatitis D virus viral life cycle and sites of investigative therapies. (1) Hepatitis D virus (HDV) virion attaches to the hepatocyte through interaction between HBsAg and NTCP; (2) HDV RNP is translocated to nucleus facilitated by HDAg; (3) HDV genome replication occurs via a “rolling cycle” mechanism; (4) HDV antigenome is transported out of the nucleus to the endoplasmic reticulum (ER); (5) HDV antigenome is translated in the ER into SHDAg and LHDAg; (6) SHDAg is transported into the nucleus; (7) SHDAg promotes HDV replication in the nucleus; (8) LHDAg undergoes prenylation prior to assembly; (9) LHDAg inhibits HDV replication in the nucleus; (10) New HDAg molecules are associated with new transcripts of genomic RNA to form new RNPs that are exported to the cytoplasm; (11)New HDV RNPs associate with HBsAg and assemble into HDV virions; and (12) Completed HDV virions are released from the hepatocyte via the trans-Golgi network.
Interferon
As IFN was thought to be able to inhibit viral nucleic acid replication, the tolerability and efficacy of IFN-α in chronic HDV patients was studied. Sixty one patients were randomized to receive IFN-α-2b three times a week (5 MU/m2for 4 mo, then 3 MU/m2for 8 mo, n = 31) vs no treatment (n = 30)[80]. A complete biochemical response was defined as the normalization of ALT or decrease of 50% from baseline and was met in 42%, 26% and 3% of treated patients at 4, 12 and 24 mo, respectively vs 7%, 7%and 0% at the same timepoints in the control group. A complete virological response was defined as negative HDV RNA and was not met in either group. A decrease in histologic portal inflammation was observed though not significant.
Forty two patients with chronic HDV infection were randomly assigned to receive either 9 million (high dose, n = 14) or 3 million units (low dose, n = 14) of recombinant IFN-α-2a three times a week for 48 wk or no treatment (control, n = 13)[81]. Biochemical response was defined as normal ALT, virologic response as the absence of HDV RNA and complete response as normal ALT and absence of HDV RNA at the end of treatment. ALT became normal in 71%, 29% (P = 0.029 compared to high) and 8% (P =0.001 compared to high) of patients in the high, low and control cohorts at 48 wk,respectively. HDV RNA was not detectable in 71%, 36% and 0% of patients in the high, low and control cohorts at 48 wk, respectively. Complete response occurred in 50%, 21% and 0% of patients in the high, low and control cohorts, respectively.Biochemical response was maintained in 50% of the patients whose ALT normalized in the high dose group and was persistent at 12 years[82]. Virological response was not sustained in any cohort but in the high dose group, there was a significant decrease in HDV RNA at the end of treatment compared with baseline (P = 0.009) and this decrease was sustained during long term follow up and two patients were able to clear HBsAg. Liver histology significantly improved in the high dose group as well in terms of interface hepatitis, lobular necrosis, portal inflammation and fibrosis stage after a mean of 11.5 years. In 4 patients in the high dose group who maintained a sustained biochemical response who had cirrhosis initially, fibrosis was absent in the last liver biopsy. Overall, long-term survival was significantly longer in the high-dose group than in the low-dose group (P = 0.019) and untreated controls (P = 0.003).
The safety and efficacy of peg-IFN-α-2b 1.5 μg/kg per week subcutaneous (SC) for 12 mo was evaluated in 14 patients with chronic HDV infection[83]. The primary endpoint was virological response with undetectable HDV RNA at the end of treatment and was met in 57% of patients and 43% of patients achieved sustained virological response (undetectable HDV RNA 6 months after end of treatment).Biochemical response was normal ALT levels at the end of treatment and was met in 36% of patients and 57% of patients achieved a sustained biochemical response(normal ALT 6 mo after end of treatment). One patient cleared HBsAg in the sustained responder group. Treatment withdrawal was not needed but dose reduction to 1 μg/kg per week occurred in 5 patients due to leukopenia. This preliminary study indicated the safety and efficacy of peg-IFN-α-2b and additional studies have been completed.
In a pilot study of ribavirin treatment in 7 patients with HDV, an oral dose of 15mg/kg daily of ribavirin was administered for 16 wk[84]. Complete biochemical response was defined as normal ALT and a complete virological response as negative serum HDV RNA. Biochemical response was not met. Significant virological response was not observed by the end of follow up and at 12 mo post-treatment. Peg-IFN-α-2b at a dose of 1.5 μg/kg for 48 wk in 2 out of 12 patients resulted in undetectable HDV RNA and normal of ALT by the end of follow up and at 24 wk post-treatment[85]. This same dose of peg-IFN-α-2b was assessed as monotherapy or in combination with ribavirin (800 mg oral daily) for 48 wk with maintenance therapy of peg-IFN-α-2b for 24 wk[86]. The primary and secondary endpoints were suppression or clearance of HDV RNA and normal ALT at end of treatment or at the end of 6 month follow up,respectively. Though not significant, the prolonged course of peg-IFN led to HDV RNA clearance and a sustained virological response in about 20% of patients in the monotherapy group. The addition of ribavarin to peg-IFN did not demonstrate added anti-HDV effects. Extending peg-IFN-α-2b therapy from 12 to 24 mo resulted in no significant advantage in virological and biochemical change[87]. Five-year therapy with peg-IFN-α-2a showed unsatisfactory results[88]. Based on these and other landmark studies, the consensus of current guidelines is treatment with peg-IFN-α for 12 mo(12-18 mo, APASL) in patients with compensated liver disease (EASL) with consideration of nucleos(t)ide analogue therapy with ongoing HBV DNA replication(AASLD, EASL) and referral to specialized centers for experimental therapies given limited efficacy of peg-IFN-α (AASLD)[15,19,20].
Interferon and nucleos(t)ide analogs
The utility of HBV nucleos(t)ide (NA) analogues as monotherapy or in combination with IFN-α therapy has been explored in chronic HDV infected patients. The efficacy of oral Lamivudine on HDV RNA, ALT, liver histology and HBsAg seroconversion was evaluated in a multicenter randomized-controlled study. Thirty one patients with chronic HDV were randomized to receive Lamivudine 100mg daily or placebo for 52 weeks then all patients received Lamividune 100mg daily for 52 weeks with 16 wk post treatment follow up. HDV RNA levels were not affected, there was a partial histological response in 26% of patients and there was no HBsAg seroconversion[89].Combination therapy with peg-IFN-α-2a, tenofovir and emtricitabine for one year in one patient with HDV showed that after 10 months of treatment, a sustained response was obtained and seroconversion of HBsAg occurred suggesting that combination therapy was more effective than IFN monotherapy[90]. Thus, more studies using mono or combination therapies were evaluated especially with adefovir.
In a large randomized treatment trial, the Hep-Net-International-Delta-Hepatitis-Intervention-Study 1 (HIDIT-I), 90 patients were treated with adefovir (ADV, 10 mg oral daily, n = 30) vs peg-IFN-α-2a (180 μg/weekly, n = 29) and placebo vs adefovir and peg-IFN-α-2a (n = 31) for 48 wk with 24 wk follow up[91]. The primary endpoint was undetectable HDV RNA and normal ALT at the end of treatment. This was met in 0, 2 and 2 patients in the ADV, peg-IFN and combination groups, respectively.HDV RNA was negative in 0%, 24% and 23% in the ADV, peg-IFN and combination groups, respectively. Twenty eight percent of patients receiving peg-IFN based treatments had negative HDV RNA at 24 wk post treatment. Zero, 2 and 10 patients developed a decline in HBsAg levels of > 1 log10IU/mL from baseline to week 48 in the ADV, peg-IFN (P = 0.01 compared to combination) and combination groups (P <0.001 compared to ADV alone), respectively.
A retrospective-prospective follow-up of the HIDIT-I trial was completed in 77 patients with a median time follow-up of 4.5 years[92]. Twenty-one, 19 and 18 patients from the ADV, peg-IFN and the combination group were included, respectively. The overall posttreatment clinical-event rate (hepatic decompensation, HCC, liver transplantation and liver-related death) was 2.5% (4.9% in patients with cirrhosis).Ten percent of peg-IFN based treated patients achieved HBsAg loss by the end of follow up. HDV RNA relapses occurred during long term follow up in nearly 50% of responder patients and the authors recommended that the term “sustained”virological response be avoided in chronic HDV infection. Univariate and multivariate analyses of HIDIT-I to identify factors associated with outcomes of peg-IFN treatment, with and without adefovir revealed that the level of HDV RNA at week 24 of treatment was associated more strongly with response to therapy and could help to identify patients who will test negative for HDV RNA 24 wk after the end of treatment[93].
In HIDIT-II, two parallel, multicenter, double-blind, randomized, controlled trials evaluated peg-IFN-α-2a (180 μg/weekly) combination therapy with oral Tenofovir Disoproxil Fumarate (TDF, 300 mg daily, n = 59) or placebo (n = 61) for 96 wk in 120 patients with chronic HDV infection[94]. The primary endpoint was negative HDV RNA at the end of treatment. Secondary endpoints included negative HDV RNA at treatment week 48 and post-treatment week 24, HBsAg loss or decline > 0.5 log10from baseline to treatment weeks 48 and 96 and normal ALT at treatment weeks 48, 96 and post-treatment week 24. Addition of TDF did not provide additional benefit for treatment endpoints as there were no significant differences between both cohorts and the authors suggest that HBV nucleotide analogous should not be recommended as standard of care at this time. However, there were other important off target findings.Ninety-six weeks of peg-IFN-α-2a was tolerated by most of the patients in this study,which also included patients with cirrhosis, and that prolonged treatment was associated with improvement in histological fibrosis scores overall. Also, the presence of liver cirrhosis was associated with an increased likelihood for an HDV RNA response at post-treatment week 24. Therefore, retreatment of patients with peg-IFN can be considered if prior treatment was tolerated and progression of disease is likely.Though peg-IFN was tolerated and showed histological improvement, this did not correlate with virological or biochemical responses and extension of treatment to 96 wk did not cause significant change in HDV RNA negativity in either group and seemed to peak at week 48 and did not prevent relapse. Therefore, if suppression of HDV RNA is the main endpoint, then treatment beyond 48 wk may not yield additional benefit.
Long-term outcome of delta hepatitis after treatment with antiviral treatment regimens was evaluated in a large, single-center cohort of 136 patients for a mean follow-up of 5.2 years[95]. Thirty-nine patients had not received treatment, 52 received IFN-α based therapy and 45 patients received treatment with NA analogues only.Clinical endpoints of hepatic decompensation, HCC, liver transplantation and liverrelated death were monitored and occurred in 55 patients. Regarding IFN-α based therapy, patients developed clinical endpoints significantly less frequently than the other two cohorts (NA, P < 0.01; untreated, P < 0.01), was independently associated with a more benign clinical long-term outcome (P = 0.04; OR = 0.25; 95% CI: 0.07-0.9),and hepatic decompensation (P < 0.01) and need for liver transplant (P < 0.01)occurred less often compared to the NA group. Though efficacy of IFN-α is not optimal, this study highlights the fact that IFN-α is necessary in those that require treatment until improved options are available.
Who to treat?
As mentioned previously, t he only recommended therapy for chronic HDV infection is IFN-α based treatment with limited efficacy and many side effects and identifying patients who warrant treatment highlight the need for specific HDV scoring systems and fibrosis markers. Current guidelines suggest treating individuals with elevated HDV-RNA levels and ALT elevation with peg-IFN-α for 12 months with consideration of NA therapy with ongoing HBV DNA replication[15,20]. In a cross-sectional study of two independent cohorts of patients including individuals from the HIDIT-II trial, anti-HDV IgM was assessed for its utility as a biomarker to determine disease activity and to predict the risk of disease progression in patients with chronic HDV infection[96]. One hundred and twenty patients were included from HIDIT-II and 78 patients from a ‘‘real-world'' validation cohort. The validation cohort received various forms of treatment: no treatment, n = 46; NA analogues, n = 16; peg-IFN, n = 3;combination NA analogues and peg-IFN, n = 13. Liver-related endpoints of hepatic decompensation (encephalopathy, variceal bleeding, ascites), liver transplantation,HCC or liver-related death were evaluated in the validation cohort. Anti-HDV IgM serum levels were tested in 102 (85%) HIDIT-II patients and 67 (85.9%) cohort patients. Negative, low, intermediate and high anti-HDV IgM levels were defined as negative, < 0.5, 0.5-2.5 and > 2.5, respectively. Though not significant, the validation cohort had more patients with intermediate (32% vs 21%) or high anti-HDV IgM levels (5% vs 0%) and in both cohorts there was a trend with histological activity.There was significant congruency between both cohorts regarding ALT and anti-HDV IgM levels (negative vs low or intermediate). In HIDIT-II patients, HBV DNA and anti-HDV IgM were inversely correlated and significant in the negative vs low or intermediate patients. There was no association between HDV RNA or HBsAg with anti-HDV IgM. Clinical long-term outcome was investigated in the validation cohort who were followed over a period of 14 years. Clinical decompensation occurred only in one anti-HDV IgM negative patient and in 26 patients with positive IgM levels (P =0.05). These findings suggest that anti-HDV IgM identified a subgroup of patients with a very mild course of liver disease who may not require urgent treatment.
INVESTIGATIONAL THERAPIES
Prohibiting entry of viral particles
Prior studies have demonstrated that myristoylation of the preS1 domain of the L envelope protein of HBsAg is required for HBV infectivity[97-99]and that interference of infectivity occurs through specific cell receptor targeting on the hepatocyte surface[100,101]. The Sodium Taurocholate Co-Transporting Polypeptide (NTCP) receptor was identified as the specific receptor for HBV and HDV[28,29]and Myrcludex B(Bulevirtide), a synthetic preS1 derived peptide, has been identified as an NTCP receptor inhibitor[99,102-104]with little effect on bile acid transport[105].
A single center, randomized, 3-arm, parallel, open-label clinical trial was performed in Russia to evaluate the effect of Myrcludex B alone or in combination with peg-IFNα-2a vs peg-IFN-α-2a alone in 24 patients with HDV infection[106]. The primary endpoint was HBsAg response at week 12 of treatment (HBsAg decline of at least 0.5 log10IU/mL at any time during the study). Secondary endpoints included the responses of HBsAg (24 wk), HDV RNA, HBV DNA and ALT at 24 and 48 wk of therapy and at 24 wk following the completion of treatment. The 3 treatment groups included the Myr cohort - 2 mg Myrcludex B SC daily for 24 wk followed by 180 μg peg-IFN-α-2a SC weekly for 48 wk; Myr-IFN cohort - 2 mg Myrcludex B SC daily plus 180 μg peg-IFN-α-2a SC weekly for 24 wk followed by peg-IFN-α-2a alone for 24 wk;IFN cohort - 180 μg peg-IFN-α-2a SC weekly for 48 wk. The results at 12 and 24 wk were reported. There was no significant change of HBsAg at 12 and 24 wk in all cohorts. At week 24, there was significant mean change from baseline in HDV RNA in 6, 7 and 6 patients and HDV RNA levels became undetectable in 2, 5 and 2 patients in the Myr, Myr-IFN and IFN cohorts, respectively. The only significant change in HBV DNA and ALT response was seen in the Myr-IFN cohort. The drug was well tolerated and there was an asymptomatic rise in plasma bile acid concentrations. At the time of publication, the trial is still ongoing as this report included data from weeks 12 and 24 of treatment.
The multicenter, open-label, randomized trial to assess the safety and efficacy of varying doses of Myrcludex B for 24 wk in combination with Tenofovir vs Tenofovir alone in patients with chronic HDV infection is completed[107]. One hundred and twenty patients were randomized to 4 treatment arms (n = 30/arm): Myrcludex B (2,5, or 10mg SC daily) and Tenofovir (245 mg daily) for 24 wk with 24 wk follow up vs Tenofovir alone for 48 wk. Myrcludex B was generally well tolerated. The primary endpoint was HDV RNA reduction by 2 log10or negativity at the end of treatment and was achieved in 46.4%, 46.8%, 76.6% and 3.3% in the 2, 5 and 10mg Myrcludex B cohorts and Tenofovir alone cohort, respectively. ALT normalization was achieved in 42.8%, 50%, 40% and 6.6%, respectively. At follow up 12 wk post-treatment, HDV RNA relapse occurred in 60%, 80% and 83% of treatment responder patients in the Myrcludex B arms, respectively.
Interim results for a phase 2, multicenter, open-label, randomized trial assessing the safety and efficacy of Myrcludex B in combination with peg-IFN-α-2a vs peg-IFN-α-2a alone in patients with compensated chronic HDV infection was made available[108].Sixty patients (n = 15/arm) were treated for 48 wk with 24 wk follow up. Treatment cohorts were as follows: peg-IFN-α-2a (180 μg weekly) vs Myrcludex B (2 mg daily) vs Myrcludex B (2 mg daily) + peg-IFN-α-2a vs Myrcludex B (5 mg daily) + peg-IFN-α-2a(180 μg weekly). Myrcludex B was generally well tolerated with an asymptomatic dose dependent increase in bile salts. The primary endpoint was undetectable HDV RNA at week 72. Secondary endpoints included undetectable HDV RNA at week 48 and ALT normalization, undetectable HBsAg or decline and combination response(decline or undetectable HDV RNA and normal ALT) at weeks 48 and 72. Median HDV RNA log10change from baseline to week 48 was -1.14, -2.84, -3.62 and -4.48,respectively. At week 48, undetectable HDV RNA was achieved in 2, 2, 9 and 6 patients, ALT normalization in 4, 10, 4 and 7 patients and combined treatment response in 2, 8, 4, 6 patients, respectively. HBsAg clearance was achieved in 0, 0, 7 and 2 patients, respectively.
In a randomized, open-label trial of daily Myrcludex B and peg-IFN-α-2a in patients with HBeAg negative chronic HDV infection, 24 patients were randomized into 3 treatment arms: Pre-treatment with Myrcludex B (2 mg daily) for 24 wk followed by peg-IFN-α-2a (180 μg weekly) for 48 wk vs combination Myrcludex B and peg-IFN-α-2a for 24 wk then peg-IFN-α-2a for 24 wk vs peg-IFN-α-2a for 48 wk and results are pending (NCT02637999). Future studies include a phase 2b study of Bulevirtide with peg-IFN-α-2a for 96 wk (NCT03852433) and a phase 3 study of Bulevirtide for 144 wk (NCT03852719) in patients with chronic HDV infection.
To begin with, he was very big and clumsy, then he had but one eye, and his teeth were long, and he stammered19 badly; nevertheless, he had an excellent opinion of himself, and mistook the Princess s cry of terror for an exclamation20 of delighted surprise
Inhibiting assembly of viral particles
As mentioned previously, HDV replication is pro moted by SHDAg and inhibited by LHDAg, which promotes assembly and export of HDV through prenylation of LHDAg. Prenylation of LHDAg promotes the interaction of LHDAg with HBsAg[53].Farnesyl is the prenyl moiety added to LHDAg by farnesyltransferase and inhibitors of farnesyltransferase have been shown to prevent the production of HDV virions[109].Lonafarnib is a farnesyl transferase inhibitor being studied in chronic HDV infection.
A proof-of-concept randomized, double-blind, placebo-controlled phase 2a trial assessed the safety, tolerability and effect of HDV RNA levels of oral Lonafarnib in chronic HDV infection[110]. Fourteen patients were randomly assigned to receive Lonafarnib 100 mg BID or placebo (Group 1) or Lonafarnib 200 mg BID or placebo(Group 2) for 28 d with follow-up for 6 months. Adverse effects including nausea,diarrhea, bloating and weight loss were notable in Group 2, but treatment discontinuation was not required. Mean log10HDV RNA levels were significantly decreased in both groups compared to placebo (Group 2 -1.54 log10IU/mL vs Group 1-0.73 log10IU/mL) and correlated with Lonafarnib concentrations. There were no significant changes noted in ALT, HBsAg or HBV DNA levels but a trend towards increased HBV DNA levels was noted in Group 2.
These findings led to a single center, phase 2, open label trial to optimize the treatment regimen of Lonafarnib with 21 patients in 7 treatment arms (Lonafarnib with and without Ritonavir in HDV-1, LOWR HDV-1)[111]. When comparing Lonafarnib 200 mg BID vs 300 mg BID monotherapy, the higher dose of Lonafarnib resulted in greater initial decreases in HDV levels though with increased gastrointestinal side effects and when combined with peg-IFN-α 180 μg weekly was intolerable resulting in discontinuation of treatment in those arms. Addition of the CYP3A4 inhibitor Ritonavir 100 mg oral daily to Lonafarnib 100 mg BID to achieve high post absorption levels of Lonafarnib without needing high doses of Lonafarnib resulted in a 2.4 and 3.2 log10reduction in HDV RNA at 4 and 8 wk of treatment,respectively. Similar results were seen with the same dose of Lonafarnib and peg-IFNα and both combinations resulted in normalization of ALT. Therapeutic flares were seen in 2/6 patients in the Lonafarnib monotherapy groups in posttreatment follow up.
In LOWR HDV-2, even lower doses of Lonafarnib proved to be effective. In this open-label, dose-optimization study, 55 patients were given combinations of Lonafarnib (25 or 50 mg BID vs 75 mg or more BID) with Ritonavir (100 mg BID), with or without peg-IFN-α (180 μg weekly) for 12, 24, or 48 wk[112]. As responses were comparable with the lower and higher dose Lonafarnib groups, the study was extended beyond 12 wk for only the 25 or 50 mg BID groups. Response was defined as HDV RNA decrease of 2 log10IU/mL or more. At week 24, 50% of patients who received Lonafarnib 50mg BID and 89% of patients who received Lonafarnib (25 or 50 mg BID) and peg-IFN-α exhibited a response. Also, at week 24, ALT levels normalized in majority of patients with combination therapy (60% with Lonafarnib 25 mg BID and 88% with Lonafarnib 50 mg BID). Therapeutic flares were also seen in several patients similar to LOWR HDV-1.
In the randomized, double-blind LOWR HDV-3 study, 21 patients received a daily oral combination of Lonafarnib (50, 75, or 100 mg) and Ritonavir (100 mg) for 24 wk[113]. Twelve patients received Lonafarnib and Ritonavir for 24 wk and 9 patients received 12 wk of placebo followed by Lonafarnib. Median decrease in HDV RNA from baseline after 12 wk of treatment was 1.6, 1.33 and 0.83 log10IU/mL in the Lonafarnib 50, 75 and 100 mg groups, respectively. Six patients achieved a decrease in serum level of HDV RNA of 2 log or more and ALT normalized in 47% of patients.
In the open-label, dose escalation LOWR HDV-4 study, 15 patients with chronic HDV infection were given Lonafarnib (50 mg BID initially, then increased to 75 mg BID after 4 wk and then increase to 100 mg BID after 1 week pending tolerability)with Ritonavir (100 mg BID) for 24 wk[114]. Sixty six percent of patients were able to tolerate dose escalation to 100 mg BID with 33% being able to maintain this dose to 24 wk. Mean decrease in HDV RNA for all patients was 1.7 log10IU/mL and ALT normalized in 53% of patients.
Finally, a multicenter, randomized, partially double-blind, phase 3 study will evaluate the safety and efficacy of Lonafarnib 50mg BID and Ritonavir 100 mg BID with and without peg-IFN-α-2a 180 μg weekly vs peg-IFN-α-2a 180 μg weekly monotherapy vs placebo for 48 wk with 24 wk follow up in patients with chronic HDV infection (Delta Liver Improvement and Virologic Response in HDV, D-LIVR)(NCT03719313). Four hundred patients will be included and will receive Entecavir or Tenofovir for at least 12 wk prior to initiating study therapy. Endpoints include virologic response (2 log10IU/mL reduction from baseline) and ALT normalization.
Preventing export of viral particles
Clinical studies in patients with treatment naive HBeAg positive chronic hepatitis B infection have shown that REP 2055 monotherapy (n = 8) for 40 wk or REP 2139-Ca monotherapy (n = 12) for 40 wk or combined treatment (n = 9/12) for 13 wk with immunotherapy (pegylated interferon or thymosin alpha 1) resulted in reduction in HBsAg and HBV DNA and development of serum anti-HBsAg antibodies[119]. In the REP 2055 cohort, HBsAg loss occurred in 3 patients during treatment and HBV DNA suppression was maintained at weeks 231 and 290 wk in 2 patients after end of treatment. In the REP 2139 cohort, 9/12 patients experienced reductions in HBsAg with 3 patients achieving HBsAg loss during monotherapy treatment. Of these 9 patients who were transitioned to combination treatment with immunotherapy,HBsAg loss was maintained in the same 3 patients and the remaining 6 patients experienced further HBsAg decline and eventually achieved HBsAg loss by the end of therapy.
In 2017, an open-label, non-randomized, phase 2 trial was published which assessed the safety and efficacy of REP 2139-Ca and peg-IFN-α-2a in patients with HBV and HDV co-infection[120]. Twelve treatment naïve, non-cirrhotic, HBeAg negative patients with HBsAg levels greater than 1000 IU/mL received 500 mg REP 2139 weekly infusions for 15 wk, then combined therapy with 250 mg REP 2139 weekly and 180μg SC peg-IFN-α-2a weekly for 15 wk and finally monotherapy with 180 μg peg-IFN-α-2a weekly for 33 wk. Primary endpoints included safety and tolerability of therapy. Secondary endpoints included HBsAg < 50 IU/mL and suppressed HBV DNA and maintenance of response during follow up. For the primary endpoints, 4 patients experienced a serious adverse event and all patients had treatment-emergent laboratory abnormalities. For the secondary endpoints, 6 patients had HBsAg levels < 50 IU/mL by the end of treatment and 5 patients maintained this by the end of 1 year follow up. Nine patients had suppressed HBV DNA (< 10 IU/mL) at the end of treatment and 7 patients were able to maintain this by the end of 1 year follow up. One patient established suppressed HBV DNA at the end of 1 year follow up. Eleven patients were HDV RNA negative during treatment, 9 remained that way at the end of treatment and 7 patients maintained this at 1 year follow up. Follow up at 1.5-2 years showed that 7/11 patients had persistent HDV RNA negativity[121].
A current study for NAPs is an open-label, randomized, active controlled, parallel comparison trial of the safety and efficacy of REP 2139-Mg (250 mg weekly) or REP 2165-Mg (REP 2139 derivative, 250 mg weekly) in combination with Tenofovir (300 mg daily) and peg-IFN-α-2a (180 μg weekly) in patients with HBeAg negative chronic hepatitis B[122]. Forty patients were treated for 24 wk with Tenofovir monotherapy before randomization into experimental (48 wk of Tenofovir, peg-IFN and REP 2139 or REP 2165) and control (48 wk of Tenofovir and peg-IFN) groups. All patients in the control group crossed over to experimental therapy at week 49 as they did not achieve a 3 log10HBsAg decline and are labeled as the adaptive control group. Interim follow up analysis of the experimental group shows that treatment was well tolerated. Eight of 10 REP 2139 and 5/10 REP 2165 patients in this group achieved functional control and normalization of liver enzymes.
Activation of host immune responses
In 2003, interferon lambda (IFN-λ), a type III interferon, was discovered[123]and was subsequently studied as an alternative treatment to IFN-α in HCV[124-127]and HBV[127,128]. Prior studies have shown that IFN-λ has similar downstream signaling pathways to IFN-α[129]but IFN-λ signaling is localized to the liver[129,130]and IFN-λ may have a protective role in the liver[131,132].
In a multicenter, randomized, open-label, phase 2 trial, the safety, tolerability and efficacy of IFN-λ was evaluated in 33 patients with chronic HDV infection (Lambda Interferon Monotherapy Study in HDV, LIMT HDV)[133]. Patients received IFN-λ 120 μg (n = 19) or 180 μg (n = 14) SC weekly for 48 wk with 24 wk follow up. Patients also received Tenofovir or Entecavir for HBV suppression. The primary endpoint was change in HDV RNA from baseline to weeks 48 and at week 72 and responders were defined as those exhibiting a 2 log10decline or below limit of quantification of HDV RNA. IFN-λ was generally well tolerated and patients that received prior treatment with IFN-α reported fewer side effects with IFN-λ. A durable virologic response was achieved in 36% in the high dose IFN-λ cohort compared to 16% in the lower dose cohort and this was maintained in both groups at 24 wk follow up. By the end of follow up, ALT normalized in 36% vs 26% in the high vs low dose cohorts,respectively.
Currently, an open label, phase 2 trial investigating the safety and efficacy of combination lonafarnib, ritonavir and IFN-λ is ongoing at the National Institutes of Health. Twenty-six patients will be treated with Lonafarnib (50 mg BID), Ritonavir(100 mg BID) and peg-IFN-λ-1a (180 μg weekly) for 24 wk with 24 wk follow up. The patients will also receive Tenofovir or Entecavir for HBV suppression. The primary safety endpoint will be drug tolerability at the prescribed dosages for the duration of treatment. The primary therapeutic endpoint is a decline of HDV RNA of 2 log10at the end of therapy. Secondary endpoints include sustained undetectable HDV RNA, loss of HBsAg, normalization of ALT, reduction in hepatic venous pressure gradient measurements and reduction in histologic inflammatory scores. To date, the study has completed enrollment and the investigators are awaiting final results.
With the increasing number of clinical trials investigating novel therapies for chronic HDV infection, it is important to determine surrogate makers to monitor treatment efficacy. A panel of experts have proposed that a ≥ 2 log10decline in HDV RNA be used as a target for initial treatment efficacy in this patient population[134].Additionally, nucleos(t)ide analogues suppressing hepatitis B should be considered as suppressing HDV can cause reactivation of HBV.
CONCLUSION
In addition to the novel treatments mentioned prior, research has been growing in identifying treatments to achieve a functional cure in chronic hepatitis B infection which could potentially be useful in the treatment of chronic hepatitis D infection[135,136]. Three areas of interest include capsid assembly modulators, immune system stimulators (toll-like receptor agonists and checkpoint inhibitors) and RNAi gene silencing. These studies in addition to those of NTCP receptor inhibitors,farnesyl transferase inhibitors, nucleic acid polymers in combination with interferon therapy will lead to further insight into the management of this devastating disease in the near future and hopefully one day a cure.
杂志排行

World Journal of Gastroenterology的其它文章
- New Era: Endoscopic treatment options in obesity-a paradigm shift
- Eosinophilic esophagitis: Current concepts in diagnosis and treatment
- Locoregional treatments for hepatocellular carcinoma: Current evidence and future directions
- Review of current diagnostic methods and advances in Helicobacter pylori diagnostics in the era of next generation sequencing
- Exploring the hepatitis C virus genome using single molecule realtime sequencing
- Surgical management of Zollinger-Ellison syndrome: Classical considerations and current controversies