Exploring the hepatitis C virus genome using single molecule realtime sequencing
2019-09-05HaruhikoTakedaTaikiYamashitaYoshihideUedaAkihiroSekine
Haruhiko Takeda, Taiki Yamashita, Yoshihide Ueda, Akihiro Sekine
Abstract Single molecular real-time (SMRT) sequencing, also called third-generation sequencing, is a novel sequencing technique capable of generating extremely long contiguous sequence reads. While conventional short-read sequencing cannot evaluate the linkage of nucleotide substitutions distant from one another, SMRT sequencing can directly demonstrate linkage of nucleotide changes over a span of more than 20 kbp, and thus can be applied to directly examine the haplotypes of viruses or bacteria whose genome structures are changing in real time. In addition, an error correction method (circular consensus sequencing) has been established and repeated sequencing of a single-molecule DNA template can result in extremely high accuracy. The advantages of long read sequencing enable accurate determination of the haplotypes of individual viral clones. SMRT sequencing has been applied in various studies of viral genomes including determination of the full-length contiguous genome sequence of hepatitis C virus(HCV), targeted deep sequencing of the HCV NS5A gene, and assessment of heterogeneity among viral populations. Recently, the emergence of multi-drug resistant HCV viruses has become a significant clinical issue and has been also demonstrated using SMRT sequencing. In this review, we introduce the novel third-generation PacBio RSII/Sequel systems, compare them with conventional next-generation sequencers, and summarize previous studies in which SMRT sequencing technology has been applied for HCV genome analysis. We also refer to another long-read sequencing platform, nanopore sequencing technology, and discuss the advantages, limitations and future perspectives in using these thirdgeneration sequencers for HCV genome analysis.
Key words: Third generation sequencing; PacBio RSII; Single molecule real-time sequencing; Hepatitis C virus; Resistance-associated substitution; Nanopore sequencer
INTRODUCTION
Anti-hepatitis C virus (HCV) therapy has drastically improved over the last decade[1].The development of oral direct-acting antivirals (DAAs) has enabled the majority of HCV-infected patients to achieve sustained virologic response (SVR)[2-13]. However,drug resistance-associated substitutions (RASs) including NS5A-P32del have been reported as one of the major causes of DAA treatment failure[14-28]. A subset of patients are difficult to treat with DAAs, such as patients with decompensated liver cirrhosis or immunosuppressed patients following liver transplantation. These patients are more likely to experience DAA treatment failure[1,29,30]. Thus, to achieve complete eradication of HCV, a more detailed understanding is needed of the hepatitis virus genome, especially of genetic alterations related to multidrug resistance.
Sequencing technology has made drastic progress in recent years[31,32]. Sanger sequencing of hepatitis viruses has been broadly applied in real-world clinical practice, mainly to predict the efficacy of antiviral therapy. Sanger sequencing can determine the major viral haplotypes present, but cannot detect low-abundance haplotypes which may have acquired RASs. By contrast, recently developed nextgeneration sequencing (NGS) instruments can generate sequence reads with much higher throughput compared to Sanger sequencing. These instruments can also detect rare nucleotide changes in variants at frequencies of less than 1% (Figure 1)[33]. Many genetic analyses of the hepatitis virus genome have been conducted using NGS over the last decade, and the utility of these methods has been validated in multiple studies worldwide. NGS has enabled detection of rare viral variants in the sera of individuals infected with HCV, analysis of the dynamics of drug-resistant variants in chronically HCV-infected patients, and even prediction of clinical outcomes such as responses to anti-HCV drugs.
Conventional NGS instruments including the Illumina Miseq, Illumina Hiseq and Ion Torrent sequencers have several serious limitations such as short read lengths(approximately 400 bp) and amplification biases. These factors restrict our ability to understand the landscape of the HCV genome[31]. One of the central limitations of conventional NGS techniques for genomic analysis of multi-drug resistant viral clones is its short-read nature. HCV has a single-stranded, 9-kbp RNA genome, and several variants associated with drug resistance are distributed over a 3-kbp region from the NS3 to the NS5A genes. Although conventional NGS can be used to evaluate the frequencies of variants present in a sample, short-read NGS of viral genomes cannot assess the linkage of nucleotide variants located at sites distant from one another in a single viral clone (haplotype). As a result, short-read NGS technologies cannot completely evaluate the population of multi-drug resistant viral clones, despite multidrug resistance being closely related to relapse during or after anti-viral therapy.
Recently, third-generation sequencing (TGS) platforms based on single molecular real-time (SMRT) technology have been developed. These platforms can generate extremely longer DNA sequences with high accuracy[31,34-36], providing us the means to obtain continuous long sequence reads from single viral clones. SMRT sequencing can be accomplished using the PacBio system (Pacific Biosciences), and several studies of viral genomes using this instrument have already been reported[32,37]. In this article, we review previous reports of HCV genetic analysis using PacBio sequencing and summarize the advantages and promise of this instrument in comparison with other NGS platforms.
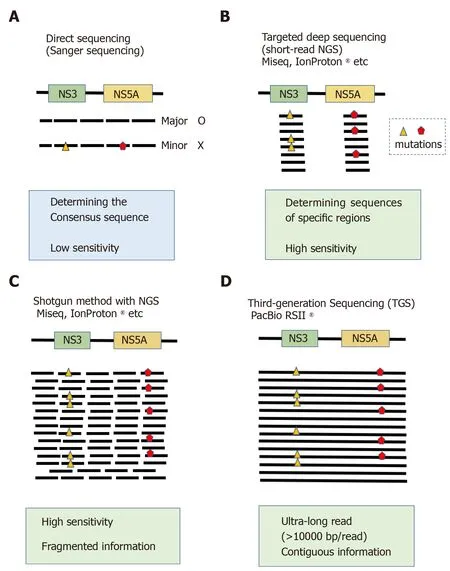
Figure 1 Comparison of sequencing platforms. A: Direct sequencing (Sanger sequencing). This conventional sequencing method determines the consensus sequence of target regions. Nucleotide variants with allele frequencies of approximately 15% can be detected; B: Targeted deep sequencing using conventional short-read next-generation sequencing (NGS) can detect low abundance variants making up approximately 1% of total mapped reads; C: When long PCR products are used as templates for conventional short-read NGS, they are first fragmented into 100-200 bp segments, ligated to sequence adapters, amplified and then sequenced. The sequenced reads are mapped to a reference sequence using the shotgun method. One of the limitations of this technique is a lack of information regarding whether two distant mutations co-exist on a single template molecule; D: Third-generation sequencing methods represented by single-molecular real-time sequencing can generate ultra-long reads of more than 10000 bp,and contiguous sequence information can be obtained. NGS: Next-generation sequencing.
SINGLE MOLECULE REAL-TIME SEQUENCING
PacBio sequencing
Recently, novel sequencing technologies capable of generating extremely long reads have been developed. As a group, these technologies have often been called TGS platforms[31]. One of the major TGS technologies is SMRT sequencing using the PacBio RSII or Sequel sequencers (Pacific Biosciences)[34,36]. SMRT sequencing is a sequencingby-synthesis technology based on real-time imaging of fluorescently-tagged nucleotides simultaneously with the synthesis along individual DNA template molecules. Because the reaction is driven by a DNA polymerase, and because single molecules are imaged in this technology, there is no degradation of signal over time.The sequencing reaction continues until the template and polymerase dissociate. The average sequencing read length from the current PacBio RSII instrument is about 12 kbp, and the newly-released PacBio Sequel sequencer can generate reads longer than 20 kbp on average. The longest sequence reads produced by the instrument exceed 50 kbp. These reads are about 200 times longer than those generated by conventional NGS instruments. Taking the advantage of ultra-long read length into consideration,SMRT sequencing has been applied to various research questions such as determination of the full-length genome sequences of bacteria, metagenome analyses of viruses or bacteria, haplotype determination of genes, transcriptome analyses of splicing variants, and determination of the full-length human genome sequence[32,38-41].
Circular consensus sequencing
The single-pass error rate for raw long reads generated by PacBio sequencing is as high as 11%-15%, with indel errors dominating. Thus, several error correction methodologies have been developed. One of the most commonly-used error correction methods is circular consensus sequencing (CCS) (Figure 2)[42,43].
The template for PacBio sequencing, called SMRTBell, is created by ligating hairpin adaptors to both ends of the prepared DNA templates, that is, double-stranded DNA molecules including PCR amplicons. The template then acts as a single-stranded closed circle loop. The enzyme initiates sequencing reaction at the specific region of hairpin adaptor (identical in all SMRTBells) and sequences the template until the polymerase loses activity. The enzyme proceeds around the hairpin on the other end of the SMRTBell and can traverse a single DNA template multiple times. Then, errorcorrected consensus reads (CCS reads) are generated using data from a single template sequenced multiple times. For example, CCS reads resulting from five polymerase passes around closed loop SMRTbells are defined as “5-pass CCS reads”.Based on the random error nature of SMRT sequencing, more passes result in higher accuracy of the consensus reads; the error rate of 5-pass CCS reads is as low as 0.1%per base and that of 10-pass CCS reads is less than 0.03%[44].
To generate 10-pass CCS reads from a 3000-bp DNA template, raw reads longer than 30000 bp should be generated by SMRT sequencing. The average length of raw reads generated by the PacBio RSII instrument is approximately 12000 bp, and typically as few as 5% of the raw reads are longer than 30000 bp. Thus, most of the sequenced reads would be excluded from the final analysis. To avoid this limitation,decreasing the template length or the pass number cutoff for analysis can be considered, although the advantages of SMRT sequencing are limited in turn.Considering that longer sequencing can generate more accurate consensus reads,improvement of sequencing cells, reagents or instruments for SMRT sequencing is expected in the future.
Nanopore sequencing
Another technology for single-molecular real-time long read sequencing is nanopore sequencing (Oxford Nanopore Technologies, Oxford, United Kingdom), which is often compared with the PacBio sequencing platform[45]. As the throughput of PacBio RS II has been somewhat limited and its running costs has been so high, many smaller laboratories have not been able to take advantage of this instrument. In 2014, the first device of a nanopore sequencer (the MinION) became available, and was immediately attractive to smaller laboratories due to its low costs and small size. Unlike other platforms, nanopore sequencers do not use base synthesis reaction in the sequencing process, which differs fundamentally from other sequencing technologies including PacBio sequencing. Instead, nanopore sequencers directly detect the sequence of the nucleotides composing a native single stranded DNA molecule via changes in electronic voltage as it passes through a protein pore.
This sequencing process generates 1D and 2D reads in which both “1D” strands can be aligned to create a consensus sequence “2D” read. The 1D raw reads have error rates of more than 10%, similar to PacBio raw reads. Although the error rates of 2D reads are somewhat improved, these are still higher than the consensus reads generated by PacBio sequencing. As a methodology for error correction of nanopore sequencing platform has not yet been established[46], its utility to detect SNVs in viral genome is limited in its current form. One group recently used nanopore sequencing to detect HCV genomes in patient sera and demonstrated that despite an error rate as high as 20%, genotypes could still be determined. Despite these limitations, nanopore sequencing is expected to be used for clinical sequencing in the future because of its low costs, USB power requirements, handheld use, and real-time processing capacity.Thus, not only improvements of instruments but also establishment of bioinformatic error correction methods is expected.
HCV GENOME SEQUENCING USING NEXT-AND THIRDGENERATION SEQUENCERS
Targeted deep sequencing of viral genomes using conventional next-generation
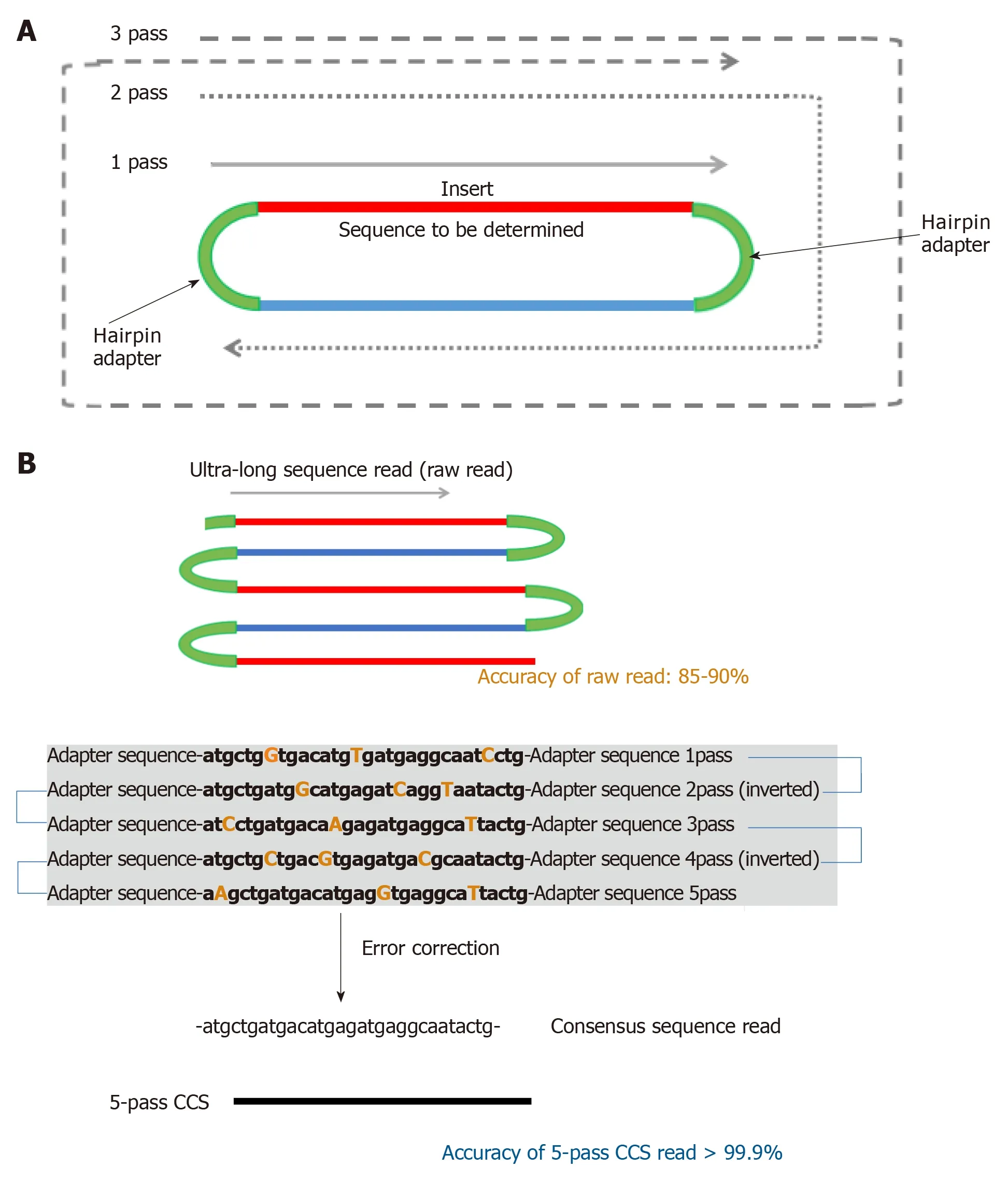
Figure 2 Generation of circular consensus sequences. A: The template for PacBio sequencing, called SMRTBell, is created by ligating hairpin adaptors to both ends of a double-stranded DNA molecule containing the sequence to be determined. This template then acts like a single-stranded closed circle. The polymerase initiates at the primer location and sequences the template until it falls off. The enzyme then proceeeds around the hairpin on the other end of the SMRTBell, and can circle around the same template multiple times; B: Scheme for generation of 5-pass circular consensus sequences (CCS) reads. Ultra-long raw reads are generated by a polymerase. Although the accuracy of the raw read is 85%-90%, error-corrected consensus reads (CCS reads) can be generated using the data from a single template sequenced multiple times. The accuracy of 5-pass CCS reads is as high as 99.9%. CCS: Circular consensus sequences.
sequencing
Conventional NGS instruments, represented by the Illumina Hiseq or ThermoFisher IonTorrent systems, are characterized by their short reads. Generally, short DNA fragments 100-400 bp in length are sequenced in massively parallel fashion[31,47]. One common NGS application is amplicon sequencing. In this application, a short region including sequences of interest (such as NS5A-aa93 in the HCV genome) is amplified by RT-PCR, followed by library generation and sequencing. Another application is based on the shotgun sequencing technique. For example, 9 kbp of the HCV genome is first amplified using long-range RT-PCR and then the long amplicons are sheared into shorter 100-200 bp fragments. Sequencing libraries are generated using the short fragments and reads are finally mapped to a reference sequence such as the fulllength HCV genome. One of the advantages of conventional NGS is its ability to generate enormous amounts of sequenced nucleotide data in less time compared with Sanger sequencing. Therefore, conventional NGS has been widely applied to examine viral quasispecies and the dynamics of viral genomes[48,49].
Using a conventional NGS method, Nasu et al[48]detected various sorts of lowabundance viral clones associated with drug resistance and characterized their dynamics in a variety of clinical settings in patients infected with HCV. This technique also enabled the discovery that various resistance-associated nucleotide alterations naturally pre-existed in treatment-naïve HCV positive patients. Sato et al[49]conducted targeted deep sequencing of the NS3 region of HCV using serum samples obtained before and after anti-HCV therapies. They compared the sequences of an approximately 450 bp segment of the HCV NS3 region and evaluated the evolution of variants resistant to interferon-based protease inhibitor therapy using phylogenetic analysis. Teraoka et al[50]compared serum samples collected from chronic HCV patients before and after oral DAA therapy and demonstrated that multidrug resistant viral clones frequently emerge at the point of treatment failure. In this manner, targeted deep sequencing of the HCV genome using conventional NGS has been widely applied and its ability to detect rare variants has been well established.After validating the reliability of variant detection by deep sequencing, targeted deep sequencing has even been applied in clinical trials of anti-HCV drugs[9].
As described above, targeted sequencing using conventional short-read NGS yields only fragmented information such as the relative frequencies of particular mutations in NS3 or NS5A. Conventional NGS cannot establish linkage between distant mutations in the NS3 to NS5A region in individual viral clones due to its short reads.Thus, conventional NGS platforms are limited in their ability to provide information regarding multi-drug resistance (Figure 2).
Targeted deep sequencing using SMRT sequencing
HCV has a single--stranded RNA genome encoding a total of 10 proteins.Nonstructural proteins including NS3, NS5A and NS5B are essential for viral replication and have been identified as the targets of DAAs. Although the therapeutic effects of DAAs are excellent, several nucleotide changes within NS regions are associated with drug resistance. In particular, nucleotide substitutions in the NS5A region are clinically important and a number of studies have used various NGS platforms for targeted deep sequencing of the NS5A region[18,19,23,24,26,50].
Targeted deep sequencing of the NS5A region of the HCV genome using the PacBio RSII platform was first reported in 2015. Bergfors et al[51]generated CCS reads from 626-bp PCR amplicons covering the NS5A region (including aa25 to aa93). The templates were prepared from 10 sera, including seven GT 1a samples, three GT 3a samples and a control plasmid.
The authors first examined the error rate by sequencing the H77 GT 1a control plasmid at NS5A aa25-95 sites and analyzing copy number, and found a mean error rate of approximately 0.05%-0.25%. The pass number of CCS reads was not noted in this analysis. They found that PacBio SMRT sequencing permitted detection of very low frequencies (as low as 0.24%) of potentially resistant HCV variants in the NS5A region. These data suggested that the detection rate of rare mutations by PacBio SMRT sequencing might be similar or even superior to that of conventional short-read sequencers.
Full-length genome determination by SMRT sequencing
Bull et al[52]described SMRT sequencing of amplicons nearly spanning the full-length HCV genome. In this report, CCS reads generated by PacBio RS II sequencing with a minimum of two passes of the full-length HCV amplicon (reads longer than 18 kb)were selected for analysis. The authors compared the sensitivity of PacBio sequencing to detect low frequency mutations with data generated by Illumina sequencing of the same amplicon. Both sequencing platforms detected all SNVs at frequencies of > 7%.However, PacBio reads only detected 4.2% of the SNVs with frequencies < 7%detected using the Illumina platform. This finding is consistent with a previous report by Jiao et al[42]. These authors conducted a benchmark study of the accuracy of CCS reads generated by the PacBio sequencer and found that the Phred-like Quality Value of 2-pass CCS reads was quite low. Thus, for accurate detection of rare SNVs, a higher pass number is needed. In order to generate 5-pass CCS reads of the full-length HCV genome (9.2 kbp), ultra-long raw reads at least 46 kb in length (five times 9.2 kb)should be sequenced. Such enormously long reads can only be rarely generated even by PacBio sequencing. Thus, accurate contiguous sequencing of the full-length HCV genome is considered a major challenge. The recently launched PacBio Sequel platform, which is reported to generate longer reads than the PacBio RSII, or improvements in sequencing polymerases might overcome this problem in the near future.
Evaluation of viral heterogeneity
To date, heterogeneity of various viral populations including HCV has been evaluated using conventional short-read sequencers. Recently, PacBio sequencing has been also applied for analysis of viral quasispecies. For example, Ho et al[53]evaluated the sequence diversity of a 1680 nucleotide-long HCV envelope genome region in individuals belonging to a cluster of sexually-transmitted cases using PacBio sequencing. Using 7-pass CCS reads, they reported an error rate of 0.37%.
We evaluated heterogeneity within the NS regions of the HCV genome in treatment-naïve HCV patients using the PacBio RS II platform[44]. For this purpose, we first performed control sequencing using a plasmid containing HCV genome as a template. We amplified its NS3/4 and NS5A regions and generated the doublestranded DNA templates for SMRT sequencing. To ensure high accuracy of sequence reads, 10-pass CCS reads were strictly selected. The average mismatch error rate of 10-pass CCS reads was 0.0287% per bp, indicating that the SMRT sequencing platform achieved extremely high accuracy sequence reads. Using these high-quality CCS reads, we applied this sequencing platform to clinical serum samples. When sequence reads were aligned to each reference sequence of HCV genome, the coverage curve showed a uniform distribution across every nucleotide position compared with the coverage curve obtained from short-read sequencing platforms. Thus, the SMRT sequencing platform can provide information on heterogeneity at each nucleotide position without positional bias (Figure 3). We performed phylogenetic analysis and found that HCV clones from chronically-infected individuals were widely distributed and that individual viral clones identified in each sample showed sequence diversity.Long-read sequencing revealed that none of the viral clones present in each individual's serum had completely identical sequences through the NS3, 4A/B, and 5A regions[44]. This finding is reasonable considering that the HCV genome is replicated by the error-prone NS5B polymerase and thus HCV clones can easily and quickly accumulate genetic mutations in their RNA genomes.
Dynamics of multi-drug resistant HCV clones
One of the most important advantages of long-read sequencing with the PacBio RSII platform is its ability to determine the haplotypes of individual viral clones (Figure 4).In the era of DAA therapy for HCV, several RASs have been identified worldwide.These RASs are mainly present in the genes encoding NS3 and NS5A. For example,NS5A-Y93H is associated with ledipasvir or daclatasvir resistance and NS3-D168V is associated with simeprevir resistance. Co-occurrence of some of these RASs in single viral clones has been reported to be associated with high rates of DAA treatment failure. Thus, evaluating the co-occurrence of RASs in NS3 and NS5A is critically important. The distance between aa168 of NS3 and aa93 of NS5A is approximately 3 kbp, and conventional short-read sequencing cannot determine linkage between RASs at NS3-aa168 and NS5A-aa93 within a single viral genome. In contrast, ultra-long read sequencing using the SMRT sequencing platform with PacBio RSII instrument can generate long contiguous sequence reads and overcome this limitation of short-read sequencers.
Long-read sequencing using the PacBio RSII can be used to evaluate not only linkage of RASs but also to analyze all synonymous nucleotide changes. Using paired serum samples collected before and after DAA treatment, we compared the haplotypes of individual viral clones and assessed the clonal evolution of HCV during DAA therapy. For this purpose, we focused on synonymous nucleotide changes linked with a given RAS such as NS5A-Y93H[44]. First, long contiguous sequences for individual viral clones present in 12 serum samples from 6 non-SVR patients (a total of more than 3000 clones) were sequenced using SMRT sequencing technology.Subsequently, all nucleotide substitutions in each viral clone before and after treatment were identified and then compared, and we found significant linkage between several synonymous nucleotide changes and major RASs. For example,several synonymous mutations were linked to NS5A-Y93H, one of the major RASs, in a subpopulation of pre-existing viral clones at baseline, and these synonymous mutations were shared by multi-drug resistant viral clones at viral breakthrough.Phylogenetic analyses revealed that pre-existing low-abundance drug-resistant clones and multi-drug resistant viral clones at viral breakthrough were genetically close each other. In addition, linkage analysis demonstrated that multiple RASs developed de novo based on pre-existing drug-resistant clones following DAA treatment in non-SVR cases. Long-read sequencing using the PacBio platform enabled us to compare the haplotypes of individual HCV clones and to estimate the origins and evolution of multi-drug resistant HCV clones during anti-HCV treatment.
PacBio long-read sequencing was also applied for analysis of multi-drug resistant clones of other virus species. Huang et al[37]examined linkage between six loci related to drug resistance of human immunodeficiency virus (HIV). They compared the drug resistance profiles of each HIV clone at two time points. The study examined a patient infected with HIV whose plasma viral load of HIV increased suddenly within one month of treatment from approximately 3000 copies/mL to approximately 30000 copies/mL. The authors found that rare viral populations with multi-drug resistant haplotypes identical to those of the major clones at the time point of relapse were already present at the pretreatment time point. They hypothesized that drug-resistant haplotypes had already existed as minor species in the viral population at the pretreatment time point and that under the selective pressure of anti-viral therapy,were quickly selected for and became dominant. Thus, long CCS reads generated by PacBio RSII sequencing can reliably provide data for HIV quasispecies-level analysis.
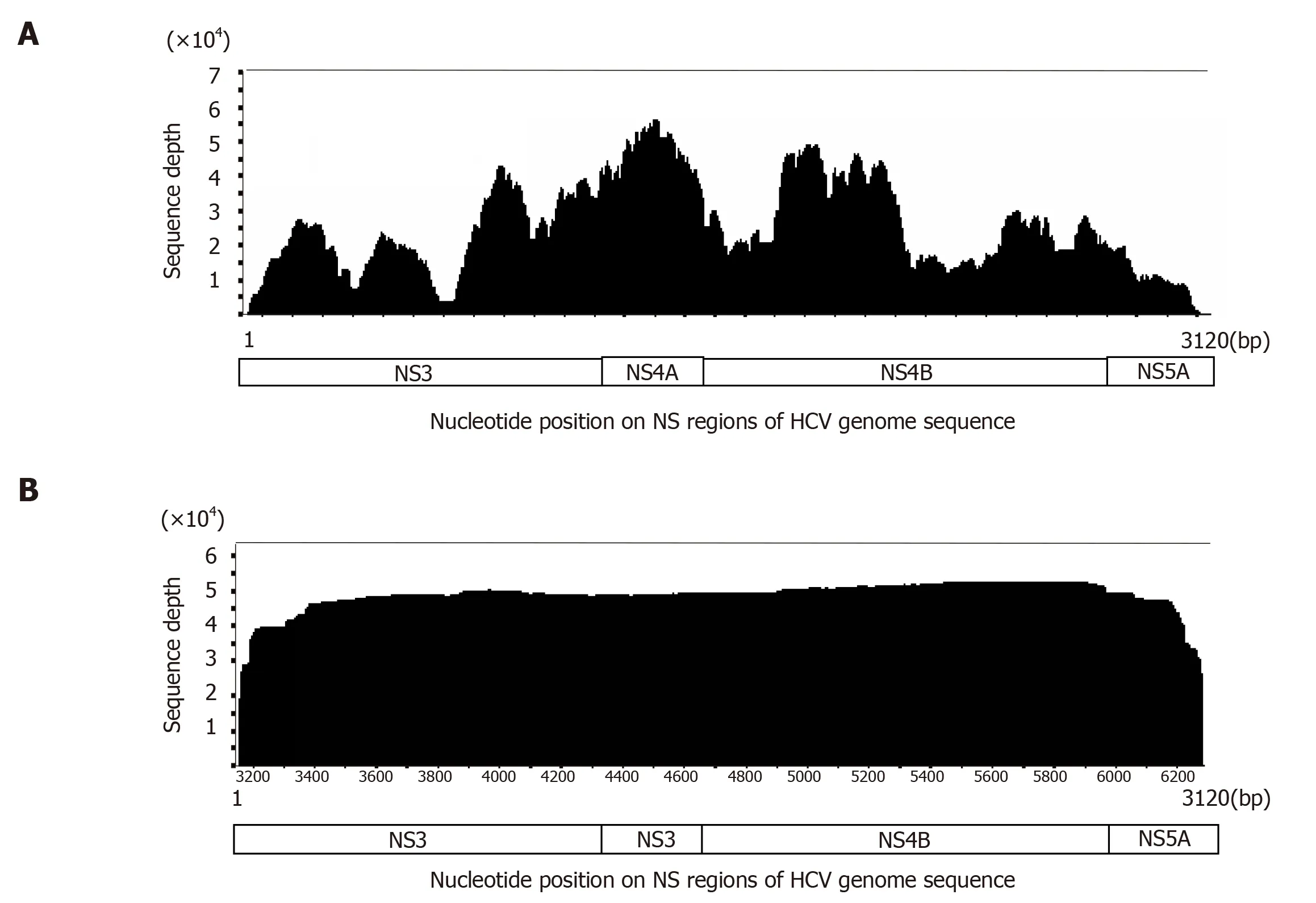
Figure 3 Comparison of coverage curves generated by short-read next-generation sequencing and long-read single-molecular real-time sequencing. A: A coverage curve generated by an IonProton sequencer. Approximately 3120 bp from the NS3 to NS5A region of the hepatitis C virus (HCV) genome from an HCVinfected patient was amplified and long-PCR products were subjected to short-read sequencing. The sequencing depth varies according to genomic location; B: When the same template was sequenced using PacBio RSII sequencer, the coverage curve demonstrates uniform coverage through the NS3 to NS5A regions. HCV:Hepatitis C virus.
CONCLUSION
SMRT long read sequencing technologies represented by the PacBio platform have opened a new era for genetic analysis of viruses. Using these sequencers, long contiguous sequences have been determined, linkage between distant SNVs can be analyzed and viral quasispecies can be analyzed in more detail than permitted by previous sequencing methods. Haplotype data generated by PacBio sequencing can be used to analyze clonal evolution of viral genomes and viral dynamics in clinical settings. Error correction methods for long reads of the HCV genome should be applicable for analysis of other viruses including hepatitis B virus, HIV or other pandemic viruses. In addition, the newly-launched PacBio Sequel System, which reportedly has seven-fold higher throughput than the RS II, is expected to further enable long-read sequencing analyses of viral genomes.
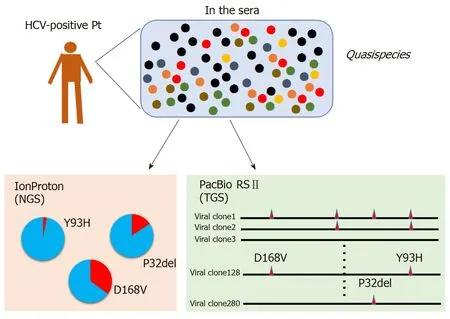
Figure 4 Comparison of short-read and long-read sequencing for analysis of viral quasispecies. Conventional short-read sequencing, such as the IonProton sequencer, generates bulk information on viral clones. However, only fragmented information can be obtained such as the frequency of viral clones bearing the NS3-D168V or NS5A-P32del variants. By contrast, PacBio RSII sequencing can determine the contiguous genome sequence of each template, permitting analysis of linkage between several nucleotide changes through the NS3 to NS5A regions for individual viral clones. TGS: Third-generation sequencing; NGS: Next-generation sequencing; HCV: Hepatitis C virus.
ACKNOWLEDGEMENTS
We thank Drs. Marusawa H, Takai A, Takahashi K, Ohtsuru S, Matsumoto T, Inuzuka T, Nakamura F and Arasawa S for helpful advice.
杂志排行

World Journal of Gastroenterology的其它文章
- New Era: Endoscopic treatment options in obesity-a paradigm shift
- Chronic hepatitis delta: A state-of-the-art review and new therapies
- Eosinophilic esophagitis: Current concepts in diagnosis and treatment
- Locoregional treatments for hepatocellular carcinoma: Current evidence and future directions
- Review of current diagnostic methods and advances in Helicobacter pylori diagnostics in the era of next generation sequencing
- Surgical management of Zollinger-Ellison syndrome: Classical considerations and current controversies