Orbitrap高分辨质谱用于保健食品中15种 非法添加减肥类药物的筛查鉴定
2019-08-29谭会洁郭常川赵艳霞张迅杰巩丽萍
谭会洁, 郭常川, 邢 晟, 赵艳霞, 张迅杰, 石 峰, 巩丽萍
(山东省食品药品检验研究院, 山东 济南 250101)
随着我国肥胖人群不断增加,减肥类药品及保健食品市场空间越来越大,无数商家竞相掘金。与此同时,市场的无序和混乱触目惊心。一些减肥类保健食品的生产厂家受利益驱动,在保健食品中非法掺入减肥类化学药,使其符合广告所宣传的治疗效果以促进销售,由于处方中没有标明添加化学药物的种类及剂量,如使用不当,将对服用者的健康和生命造成危害。为有效打击这种制假掺伪的不法行为,有必要建立快速、专属的方法鉴定保健食品中非法掺入的此类药物。
与减肥功效相关的药物主要包括食欲抑制剂(如西布曲明、芬氟拉明)、中枢兴奋剂(咖啡因、麻黄碱等)、泻药(大黄素等)、利尿剂(呋塞米)等[1]。目前已报道的用于检测非法添加减肥类药物的分析方法主要包括薄层色谱联用显微红外光谱法[2]、薄层色谱联用原位拉曼光谱法[3]、高效液相色谱法(HPLC)[4,5]、液相色谱-质谱联用法(LC-MS)[1,6-13]、液相色谱-高分辨质谱联用法(LC-HRMS)[14,15]、流动注射-质谱联用法[16]等。LC-MS由于其具有快速简便、灵敏度高、选择性好的优势,是目前国内外非法添加检验领域的最常用方法[17-19]。已报道的LC-MS以低分辨质谱法为主,也有两篇高分辨质谱法的文献采用了飞行时间质谱(TOF)[14,15]。作为高分辨质谱两种不同类型的质量分析器,相对于TOF, Orbitrap由于其质谱扫描分辨率的优势(最高可达1 000 000 FWHM[20]),能够大幅提高筛查定性能力,保证结果更加真实可靠,同时具有媲美三重四极杆的定量性能。
本研究根据近年来国内外相关文献[1-16]及多年的日常非法添加检验,确定了西布曲明、酚酞等15种非法添加减肥类化合物作为分析物,旨在开发一种简便可靠的超高效液相色谱-Orbitrap高分辨质谱联用(UHPLC-Orbitrap HRMS)方法,同时构建TraceFinder筛查数据库和自动筛查方法,用于保健食品中15种非法添加减肥类药物的高精度筛查、鉴定和定量,为日常的减肥类非法添加检测工作提供更可靠的技术支持。
1 实验部分
1.1 仪器、试剂与材料
Thermo Q ExactivePlusTM超高效液相色谱-质谱联用系统包括Ultimate 3000液相泵、自动进样器、柱温箱以及Orbitrap高分辨质谱(Thermo,德国); XCalibur 4.0软件(Thermo,美国),用于质谱仪控制和数据处理;Mettler XS205电子天平(梅特勒,瑞士); Sigma 3K15高速冷冻离心机(Sigma,德国); KQ-300 GDV温控超声仪(昆山市超声仪器有限公司)。
15种减肥类对照品均购自中检院(含量均>98.0%); HPLC级甲醇(纯度99.9%)、乙腈(纯度99.9%)以及超纯水均购自Fisher公司(美国), HPLC级乙酸铵(纯度97%)购自ACS恩科化学公司(美国);样品信息:29批保健食品(全部为胶囊剂)来自山东省食品药品监督管理局或山东省各市、县食品药品监督管理局监督抽样。随机挑选未检出15种减肥类成分的10批样品,混合均匀,作为阴性样品基质。10批阴性样品编号分别为Sample 4、Sample 7、Sample 8、Sample 10、Sample 13、Sample 16、Sample 19、Sample 20、Sample 22和Sample 29。
1.2 实验条件
1.2.1对照品溶液的制备
分别取麻黄碱、克仑特罗、盐酸芬氟拉明、盐酸西布曲明对照品(A组)适量,精密称定,分别用甲醇溶解并配成500 mg/L的对照品储备液,各取适量,以甲醇稀释为100 mg/L的混合对照品溶液I;分别取咖啡因、呋塞米、氯噻酮、苯佐卡因、布美他尼、依他尼酸、酚酞、苄氟噻嗪、大黄素、螺内酯、奥利司他对照品(B组)适量,精密称定,分别用甲醇溶解并配成500 mg/L的对照品储备液,各取适量,以甲醇稀释为100 mg/L的混合对照品溶液II。对照品储备液、混合对照品溶液I和II均放置于-20 ℃冰箱保存。临用前,恢复至室温使用。
分别取混合对照品溶液I和II适量,以50%(v/v)甲醇水溶液逐级稀释为系列对照品标准溶液(A组化合物质量浓度为1.0、5.0、10、20、50、100、200 μg/L, B组化合物质量浓度为5.0、25、50、100、250、500、1 000 μg/L)。
1.2.2供试品溶液的制备
取供试品适量,研细后称取约0.5 g,精密称定,置于50 mL具塞离心管中,加入甲醇20 mL,涡旋1 min混匀,20 ℃恒温超声提取15 min,于10 000 r/min、4 ℃条件下离心5 min,上清液过0.22 μm微孔滤膜,取续滤液即得供试品溶液。若测得化合物浓度超过定量上限,则以50%(v/v)甲醇水溶液作为稀释液,将供试品溶液稀释后再进行测定。
1.2.3色谱条件
色谱柱为Thermo Hypersil GOLD C18(100 mm×2.1 mm, 3 μm),流动相:10 mmol/L的乙酸铵溶液(A)-乙腈(B),梯度洗脱程序:0~0.5 min, 5%B; 0.5~20.0 min, 5%B~90%B; 20.0~22.5 min, 90%B; 22.5~22.6 min, 90%B~5%B; 22.6~25.0 min, 5%B。流速:300 μL/min;柱温:45 ℃;自动进样器温度:20 ℃;进样量:5 μL。

表 1 15种减肥类化合物的Orbitrap HRMS数据库Table 1 Orbitrap HRMS database of the 15 compounds
1.2.4质谱条件
Q ExactiveTM质谱系统配有HESI源,采用正负离子同时扫描模式,喷雾电压为3.0 kV(正离子模式)或2.5 kV(负离子模式),毛细管温度和喷雾温度分别为350 ℃和250 ℃。鞘气和辅助气流速分别设为40和15 arb, S-lens RF电压为50 V。质谱扫描方式为Full MS/dd-MS2模式。Full MS(一级质谱全扫描)分辨率设为70 000 FWHM,质谱扫描范围为m/z100~1 000,自动增益控制(AGC)、最大注入时间(IT)分别设为1.0e6和100 ms; dd-MS2(数据依赖的二级质谱扫描)分辨率设为17 500 FWHM, AGC设为2.0e5, IT设为50 ms,分离窗口设为m/z1.0,强度阈值(intensity threshold)设为4.0e4,同位素排除设为“on”,动态排除设为10.0 s。各化合物的步进归一化碰撞能量(NCE)统一设为20、40、60。表1列出了15种化合物的分子式、保留时间、分子离子准确质量、碎片离子准确质量等信息。
1.3 软件
使用TraceFinder 3.3软件(Thermo,美国)进行筛查定性,使用XCalibur 4.0软件(Thermo,美国)进行定量分析,使用Excel (Microsoft,美国)进行必要的计算。
2 结果与讨论
2.1 实验条件考察
为简化操作步骤并提取得到尽可能多的化合物,本研究对超声提取条件进行了优化。分别考察了不同提取溶剂(甲醇、乙腈、50%(v/v)甲醇水溶液、50%(v/v)乙腈水溶液)和不同提取时间(10、15、30 min)的提取效果。结果表明,甲醇作为提取溶剂提取效率最高,而超声提取15 min和30 min均可达到较高的提取效率。考虑到节省样品预处理时间,本研究选择甲醇作为提取溶剂、超声提取15 min。
分别以50%(v/v)甲醇水溶液稀释对照品储备液,得到10 mg/L的15种化合物标准溶液,分别使用单针进样的方式注入质谱仪,在正、负离子模式下进行全扫描,优化质谱条件(质谱扫描极性、分子离子准确质量、喷雾电压、毛细管温度和喷雾温度、鞘气和辅助气流速等),输入至方法设置中。在建立的LC-MS联用方法中,质谱扫描方式选择正负离子切换Full MS/dd-MS2模式,此模式包含一级全扫描和数据依赖的二级扫描,得到高精度一级质谱和二级质谱。其中,高分辨一级质谱图用于化合物的筛查、鉴定和定量,二级质谱图用于化合物确认。
为选择最佳流动相,本研究考察对比了甲醇-水和乙腈-水混合体系,并在水相中分别添加了0.1%(v/v)甲酸水溶液、10 mmol/L甲酸铵、10 mmol/L乙酸铵等。结果表明,乙腈-10 mmol/L乙酸铵流动相体系可获得最佳的色谱峰形和分离效果,且正离子和负离子模式的不同化合物均能获得较高的质谱响应。因此,本研究选择乙腈-10 mmol/L乙酸铵溶液作为流动相体系,并采用梯度洗脱程序。

图 1 15种减肥类化合物的提取离子色谱图Fig. 1 Extracted ion chromatograms of the 15 weight loss compounds
2.2 方法学考察
2.2.1专属性
取阴性样品,按1.2.2节方法处理得到空白基质溶液,进样分析。空白基质溶液的提取离子色谱图(extracted ion chromatograms, XICs)中,各化合物保留时间处没有干扰峰出现或干扰峰很小(低于定量限的10%),方法专属性强。
取对照品标准溶液(A组化合物浓度为5.0 μg/L, B组化合物浓度为25 μg/L)进样分析,记录色谱图(见图1)。15种减肥类化合物可在25 min内完成色谱分离和洗脱,峰形好,灵敏度高。代表性化合物----芬氟拉明的二级质谱图及裂解机理如图2和图3所示,二级质谱碎片离子有助于确证化合物的存在,裂解机理阐明了主要碎片离子可能的化学结构及裂解途径。
2.2.2基质效应
分别制备空白基质加标溶液和对照品标准溶液(A组化合物浓度约为20 μg/L, B组化合物浓度约为100 μg/L),分别进样,计算二者响应值百分比,即为基质效应[21]。15种化合物的基质效应在87.9%~105.4%范围内,证明空白基质对化合物响应影响很小。
2.2.3线性范围和灵敏度
取系列标准溶液进样测定。以峰面积作为y,标准溶液质量浓度作为x,拟合校正曲线,建立回归方程,计算相关系数(r)。15种化合物的峰面积与质量浓度的线性关系良好,r均大于0.998,线性范围宽,可满足准确定量的需求。以标准曲线最低点作为定量限(S/N≥10)。结果见表2。

图 2 芬氟拉明的二级质谱图及碎片离子结构推断Fig. 2 MS2 spectrum of fenfluramine with fragment ion structures tentatively assigned
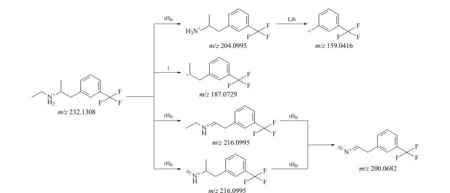
图 3 芬氟拉明的二级碎片裂解机理Fig. 3 Fragmentation mechanism of fenfluramine
2.2.4回收率和精密度
精密称取阴性供试品0.50 g至50 mL具塞离心管中,分别加入20 mg/L的A组混合对照品溶液20 μL、20 mg/L的B组混合对照品溶液100 μL,再加入20 mL甲醇,按照1.2.2节所述方法处理得到供试品溶液,其中A组化合物质量浓度约为20 μg/L, B组化合物浓度约为100 μg/L。平行测定6次,计算回收率平均值和精密度(RSD),数据见表3。得到15种减肥类化合物的回收率范围为79.7%~95.4%,精密度在3.3%~8.7%之间。
2.2.5稳定性
考察了100 μg/L标准溶液在自动进样器中放置1、2、4、8、12、24、48 h的稳定性,15种化合物在上述时间下的稳定性为 97.8%±2.5%(平均值±RSD),说明该条件下样品稳定性良好。
2.3 筛查数据库的开发与应用
本研究通过TraceFinder 3.3软件实现了快速自动筛查。首先,建立筛查数据库和筛查方法。通过对照品溶液的UHPLC-HRMS进样分析可获得15种减肥的保留时间、一级质谱和二级质谱准确质量,据此建立15种减肥类化合物的筛查数据库,如表1所示。在数据库中保存化合物的特征信息,如化合物名称、分子式、可能的加合物(如[M+H]+、[M-H]-等)、保留时间、分子离子准确质量、碎片离子准确质量等。然后设定筛查的各项参数,建立筛查方法。以分子离子和保留时间作为鉴定依据,碎片离子和同位素分布作为确证依据。峰面积阈值设为10 000 000,分子离子准确质量和碎片离子准确质量偏差阈值均设为5×10-6(5 ppm),保留时间窗口设定为15 s,用于确证的碎片离子数量至少为2,同位素分布的拟合阈值设为90%,质量偏差阈值设为5×10-6(5 ppm),同位素离子强度偏差阈值设为10%。
将UHPLC-HRMS采集的所有样品原始数据(raw格式)直接导入TraceFinder软件,以建立的筛查数据库和筛查方法进行自动筛查。软件会把仪器采集的样品信息与筛查数据库、筛查方法和接受标准进行自动比对。如果样品中某个离子的4个筛查选项(即分子离子准确质量、保留时间、碎片离子准确质量及同位素分布)均符合筛查标准(通过标准则绿灯亮起),则判定样品为阳性。图4~6给出了代表性样品Sample 3的提取离子色谱图、一级全扫描质谱图、同位素分布比对图及二级质谱比对图,图中显示了Sample 3的[M+H]+232.130 8离子实测和理论保留时间偏差(0.03 min,即1.8 s)、分子离子准确质量与理论值偏差(-1.256 1×10-6(-1.256 1 ppm))、碎片离子准确质量与理论值偏差(均小于2×10-6(2 ppm))及同位素分布(评分为100%)均在筛查方法设定的接受标准之内。4个筛查选项标记均为绿灯,Flag标记也为绿灯,表明软件判定Sample 3检出了芬氟拉明。因此,使用已建立的数据库和筛查方法进行高度可靠的快速自动筛查,可以将假阳性和假阴性样品的风险降至最低。
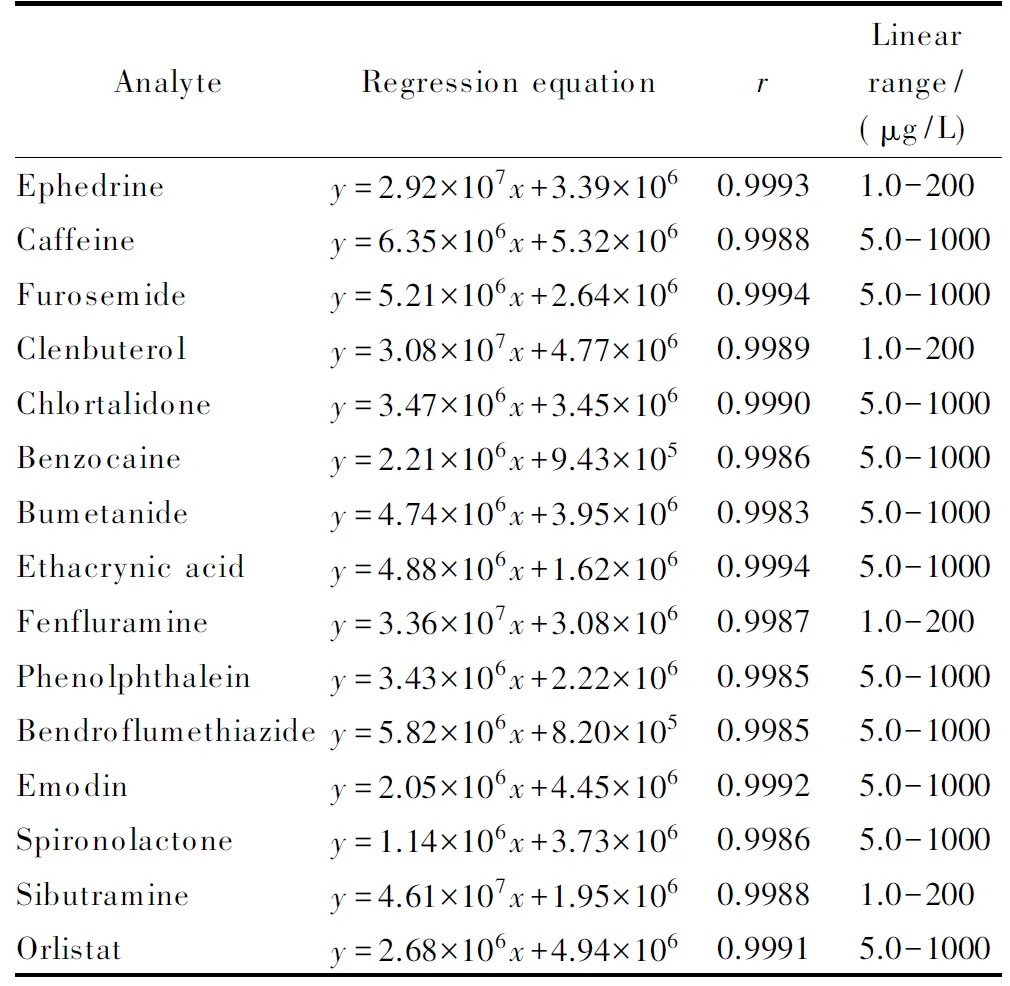
表 2 15种化合物的回归方程、相关系数和线性范围Table 2 Regression equations, correlation coefficients (r), and linear ranges of the 15 compounds
y: peak area;x: mass concentration, μg/L.

表 3 减肥类化合物的回收率和精密度(n=6)Table 3 Recoveries and precisions of the 15 compounds (n=6)

图 4 代表性阳性样品的提取离子色谱图及一级全扫描质谱图Fig. 4 Extracted ion chromatograms (XICs) and full scan mass spectrum of a representative positive sample

图 5 代表性阳性样品中芬氟拉明的同位素分布及TraceFinder比对Fig. 5 Isotope pattern and the TraceFinder fit of a representative positive sample

图 6 代表性阳性样品中芬氟拉明的二级质谱图及TraceFinder比对Fig. 6 MS2 spectra and the TraceFinder fit of a representative positive sample
2.4 实际样品检测
将建立的UHPLC-Orbitrap HRMS分析方法用于29批保健食品样品中非法添加减肥类药物的检测,将得到的raw原始数据文件导入TraceFinder 3.3筛查软件,根据TraceFinder中建好的筛查数据库和筛查方法进行快速自动筛查,并使用XCalibur软件对阳性样品进行定量分析。
在总计29批次样品中检出6批阳性样品,阳性率为20.7%。检测到的非法添加物涉及盐酸西布曲明、酚酞、盐酸芬氟拉明、依他尼酸,非法添加物含量(见表4)差异很大,有的甚至远超临床用量。此外,在阳性样品中还发现了几种化合物同时添加的现象,如Sample 14同时添加了盐酸芬氟拉明与依他尼酸。这些结果表明了非法添加的随意性及不可预料性,对消费者健康具有较大危害。

表 4 阳性样品检出成分及含量Table 4 Detected compounds and their contents in positive samples
3 结论
本研究建立了检测保健食品中非法添加减肥类药物的液相色谱-高分辨质谱联用法,采用TraceFinder软件构建了Orbitrap高分辨质谱数据库,建立了筛查方法,将液相色谱-质谱联用仪采集的数据导入TraceFinder软件进行快速、自动筛查分析。阳性样品可使用XCalibur软件进行定量。本方法灵敏度高、选择性好,筛查分析结果准确可靠,自动化程度高,可满足同时定性和定量的需求,适用于保健食品中减肥类药物的非法添加检测,已用于日常检测工作中,为监管减肥类非法添加提供了有力的技术支持。
