类铝钛离子的能级与跃迁特性计算
2019-08-27梅茂飞桑萃萃杨家敏
胡 峰, 孙 言, 梅茂飞, 桑萃萃, 杨家敏
(1. 徐州工程学院 数学与物理科学学院, 江苏 徐州 221018; 2. 兰州理工大学 理学院, 甘肃 兰州 730050;3. 中国工程物理研究院 激光聚变研究中心, 四川 绵阳 621900)
高电离态离子性质的研究在高电离态原子、X射线、惯性约束聚变、磁约束聚变、天体和等离子体物理研究中有着重要的意义和广泛的应用价值.等离子体中高电离态离子光谱一直作为等离子状态诊断的重要工具被广泛应用,因此提供精确的光谱数据显得尤为重要.Ti元素目前作为示踪元素在激光等离子体中有重要的应用,可以将Ti和Cr合金片埋于黑腔内壁上,通过测量2种元素离子的谱线强度,推导出等离子体的温度和密度关系[1].类铝Ti离子光谱的实验与理论研究较少,Froese等[2]用多组态Hartree-Fock方法(MCHF)计算了Ti X离子的能级、能级寿命与跃迁几率;Santana等[3]用Multireference Mller-Plesset perturbation(MPPT)方法研究了Ti X离子的特性;随后,Singh等[4]用同样的方法研究了Ti X(增加了相关组态);Aggarwal等[5]用基于多组态Dirac-Fock方法(MCDF)的程序包GRASP和组态相互作用方法的程序包Flexible Atomic code (FAC)[6]研究了Ti X相关能级、波长等相关数据.实验只有Träbert等[7]用重离子存储环研究了Ti X的能级寿命.
上述提到理论计算结果与实验结果存在一定的差距,并不能够获取较为精确的实验结果.因此本文利用多组态Dirac-Hartree-Fock方法(MCDHF)的程序包GRASP2K[8-9],通过考虑电子关联效应,去获得更加精确的数据和理解差距产生的原因.
1 计算方法
1.1 波函数和能级本文所用的MCDHF方法在文献[10-12]中已有详细描述,在该方法中,一个核电荷数为Z、具有N个电子的原子或离子体系的Dirac-Coulomb 哈密顿量为(原子单位)
(1)
用Hi表示第i个电子的Dirac哈密顿量
(2)

(3)
其中,n是主量子数,m是磁量子数,k为Dirac量子数,Pnk(r)和Qnk(r)分别为相对论径向波函数的大小分量,χkm为自旋函数.
N电子体系的组态波函数|Γr(PJM)〉是所有单电子旋-轨波函数组成的N阶Slater行列式波函数|Ψp〉的线性组合,即
(4)
在MCDF 方法中,任一原子态α的波函数|α(PJM)〉由具有相同P、J和M量子数的组态波函数,|Γr(PJM)〉线性组合而成,即
(5)
其中,nc是组态波函数的个数,Cr(α)为组态混合系数.
对角化由原子波函数(5)式构造的哈密顿矩阵,则可得到相关原子态的能量和组态混合系数.对于其他高阶效应,如Breit修正和主要的量子动力学效应,可作为微扰处理.
1.2 电子关联效应在具体计算过程中,主要是通过逐渐增加基矢数目来考虑更多的组态相互作用,直至得到收敛的结果.可以把组态相互作用分为2类:价电子之间的相互作用 (Valence-Valence Correlation)称为VV关联效应,原子实内的电子与价电子之间的相互作用( Core-Valence Correlation)称为CV关联效应.一般来说,只考虑VV关联效应就能得到比较精确的结果,但是对于电子数目比较多的体系,如果要得到比较精确的结果,CV关联效应是必须考虑的[13],在本文计算中为了得到比较精确的结果,对这2种关联效应都进行了考虑.
1.3 计算步骤文献[8-9]已经详细介绍了电子关联作用,这里只简单地介绍主要部分.为了更好解释如何考虑电子关联效应,考虑以下2个步骤:
1) 运行Dirac-Fock初步得到1s22s22p63s2的径向波函数.在这一步里,选用Thomas-Fermi模型势作为计算的初始波函数.在自洽场计算中,对所有的轨道都进行了优化.
2) 设置n=1-2为闭壳层,也就是说主量子数为1和2的轨道上的电子是封闭的,既不允许这些电子向其他轨道跃迁也不允许其他轨道的电子跃迁到这2个轨道.允许n=3的轨道一个电子可以跃迁到n=4,5,6,7轨道上,且任意分布,这样就考虑了VV关联效应,其扩展的轨道形式为
1s22s22p6nln′l′.
而对于CV关联效应来说,此时考虑2种情况,一种是2p轨道上一个电子不受限制,或者另一种是2s上一个电子不受限制.要求对于一个双激发,一个电子必须来自原子实中的2p(2s)轨道,另一个电子必须来自价电子.这样原子实n=2轨道电子和价电子之间的CV关联效应就考虑进去了,其扩展的轨道形式为:
1s22snl2p6n′l′n″l″, 1s22s22p5nln′l′n″l″ .
2 结果与讨论
为了更好的解释VV和CV电子关联效应的影响,在表1中列出了2种方法计算出的3s23p和3s3p2的能级,结果包含了量子电动力学效应和Breit修正,其中量子电动力学效应考虑自能和真空极化2种修正.实验值采用美国国家标准局原子与分子数据库的数据作为参考[14].从表1可以看出,考虑了电子关联效应之后,当前计算精度有了较大的提高,与实验值更加符合.例如能级9和10,在只考虑VV关联效应时,与实验值的偏差在1.207%和1.228%;而考虑CV关联效应后,偏差缩小到了0.095%和0.084%.为了进一步展示考虑CV关联效应带来的影响,在图1中对比了不同理论值与实验值的差异.

图 1 理论计算值与实验值的比较
其中GRASP1和GRASP2的数据是来自文献[5],基于MCDF方法分别考虑530个能级和1 387个能级给出的计算结果,FAC1和FAC2同样来自文献[5],是基于组态相互作用分别考虑1 387个能级和12 139个能级给出的计算结果.MCHF的计算结果来自文献[2].从图1可以看出,当前计算结果与实验值的最大偏差只有0.603%,而GRASP和FAC的偏差达到了2.180%和1.751%.结果表明,考虑电子关联效应,尤其是内壳层的电子,对于计算结果影响是比较大的.

表 1 类铝钛离子3s23p和3s3p2能级
注:a 偏差=|VV值-实验值|/实验值*100;b偏差=|CV值-实验值|/实验值*100.
同时可以看出随着考虑能级数量的增加,计算结果越接近实验值,但是通过对比发现理论计算的3s23p2P3/2的值与实验值都有较大的差距,当前计算结果和文献[4]的结果都显示3s23p2P3/2组态混合贡献为0.98,按照组态混合的贡献应该得到很好的结果,之所以出现差距原因可能是由于早期计算结果没有考虑能级混合的影响.
等离子体中发射的谱线可以用来诊断电子温度和密度[1],因此准确的波长对于研究等离子体的状态是必不可少的.同时为了验证当前计算的可信性,在表2中给出了类铝钛离子跃迁波长的理论计算值和实验值,其中表2中的本文计算结果是考虑电子关联也即CV关联效应的结果.同时表2也列出了文献[2]的MCHF计算结果和文献[4]的组态相互作用计算结果,实验值同样选取NIST给出的参考值[14].从表2可以看出,当前本文计算值与文献[14]实验值符合较好,比值在0.995~1.007,这样的不确定度与实验可能产生的偏差相一致,从而验证了当前计算的精确性.同时看出当前计算值与实验值的偏差在-0.035~0.467 nm,要好于文献[2]的-0.422~0.607 nm.文献[4]的计算结果部分优于本文,但是计算结果不够完整,考虑跃迁数量明显偏少.
对于从能级i态到j态跃迁的电偶极振子强度(fij)和跃迁几率(Aji)的关系可以用下面的公式来定义[15]:
(6)
其中,m和e分别是电子质量和电荷,c是光速,λji是跃迁波长(nm),ωi和ωj分别是下能级i和上能级j的权重.对于电偶极跃迁来说,跃迁几率和振子强度可以给出如下的形式:
(7)
其中S是线强度,单位为原子单位a.u..
表3给出了类铝钛离子3s23p—3s3p2跃迁几率,同时也给出了来自文献[2,4-5]不同计算方法计算结果.其中文献[2,4]都采用了2种不同方法计算了跃迁几率,可以看出其计算值与本文计算值、实验值符合很好.需要说明的是文献[4]报道的
3s23p2P3/2— 3s3p22P1/2
跃迁几率为8.3×109s-1,该值明显偏小.本文计算的
3s23p2P3/2— 3s3p22D3/2
跃迁几率为1.2×108s-1,与实验值的偏差达到了26%,但是与文献[2,4]的计算值相当,说明该跃迁几率的实验值是有争议的,需要在今后的实验中做进一步的测量.
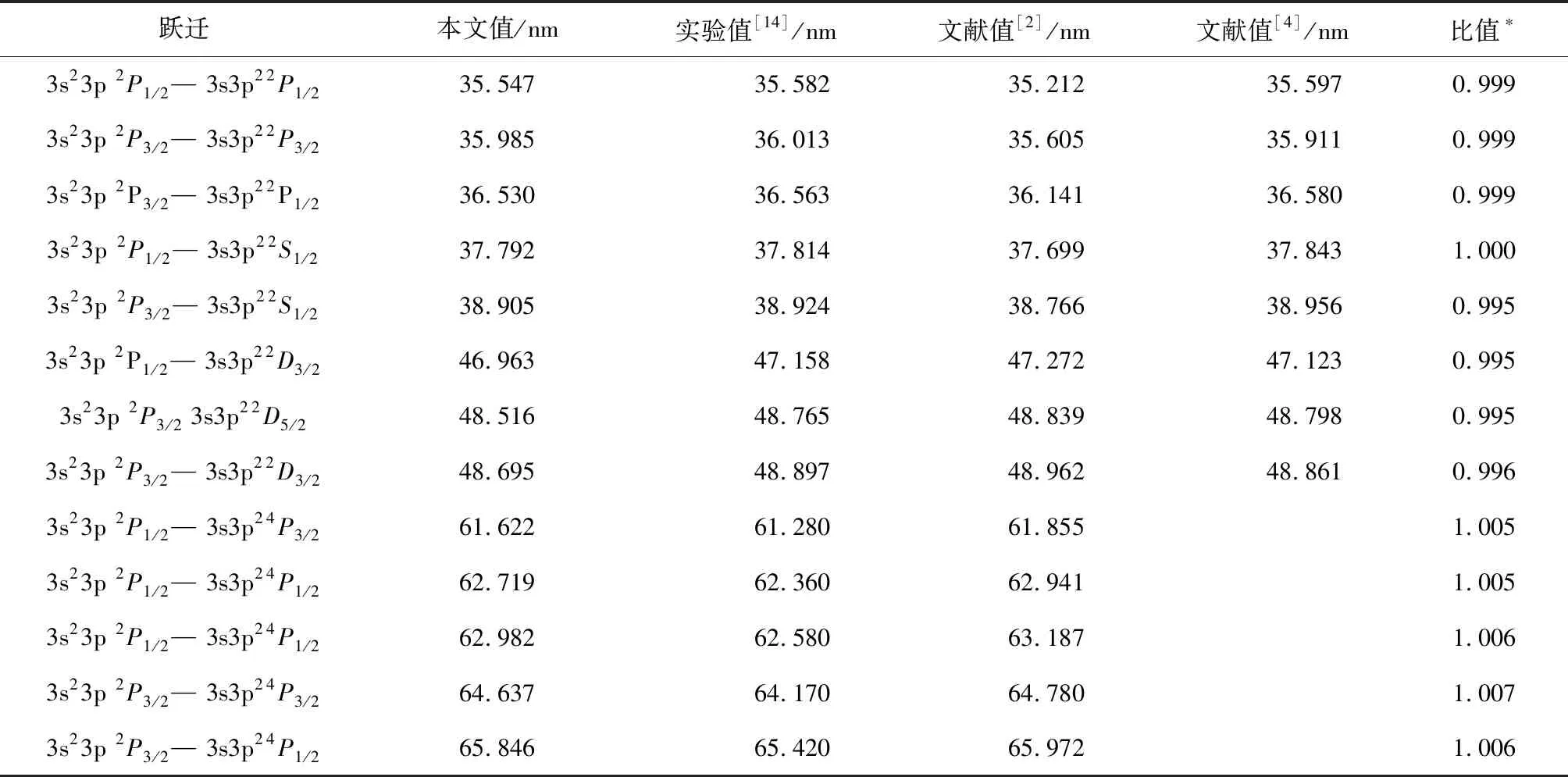
表 2 类铝钛离子的波长
注:*比值=本文值/实验值[14].

表 3 类铝钛离子3s23p—3s3p2跃迁几率
表4给出了类铝钛离子3s23p—3s3p2振子强度,同时也给出了来自文献[2,5]不同计算方法计算结果.需要说明的是,振子强度不是一个可以直接测量值,可以通过(6)式进行换算,因此为了验证当前振子强度计算的精确度,引入一个代表原子结构计算中总的长度规范下的振子强度(fgl)与速度规范下的振子强度(fgv)的比值,即fgl/fgv.该比值代表的是原子结构计算中的2种不同规范(长度规范和速度规范),一般认为2种规范的比值越接近于1,表明计算结果越精确[16].从表4可以看出当前计算的比值在0.97~1.07区间,明显好于文献[4]的0.88~1.66,表明当前振子强度的计算是真实可信的.为了丰富类铝钛离子3s23p—3s3p2的跃迁数据,在表5和6中分别给出了电四极、磁偶极与磁四极的跃迁数据,因为当前并没有实验数据作为比对,因此本文的计算结果可以作为辨识这些谱线的参考.

表 4 类铝钛离子3s23p—3s3p2振子强度
注:a比值为文献值[4] 中的fgl/fgv;b比值为本文值fgl/fgv.
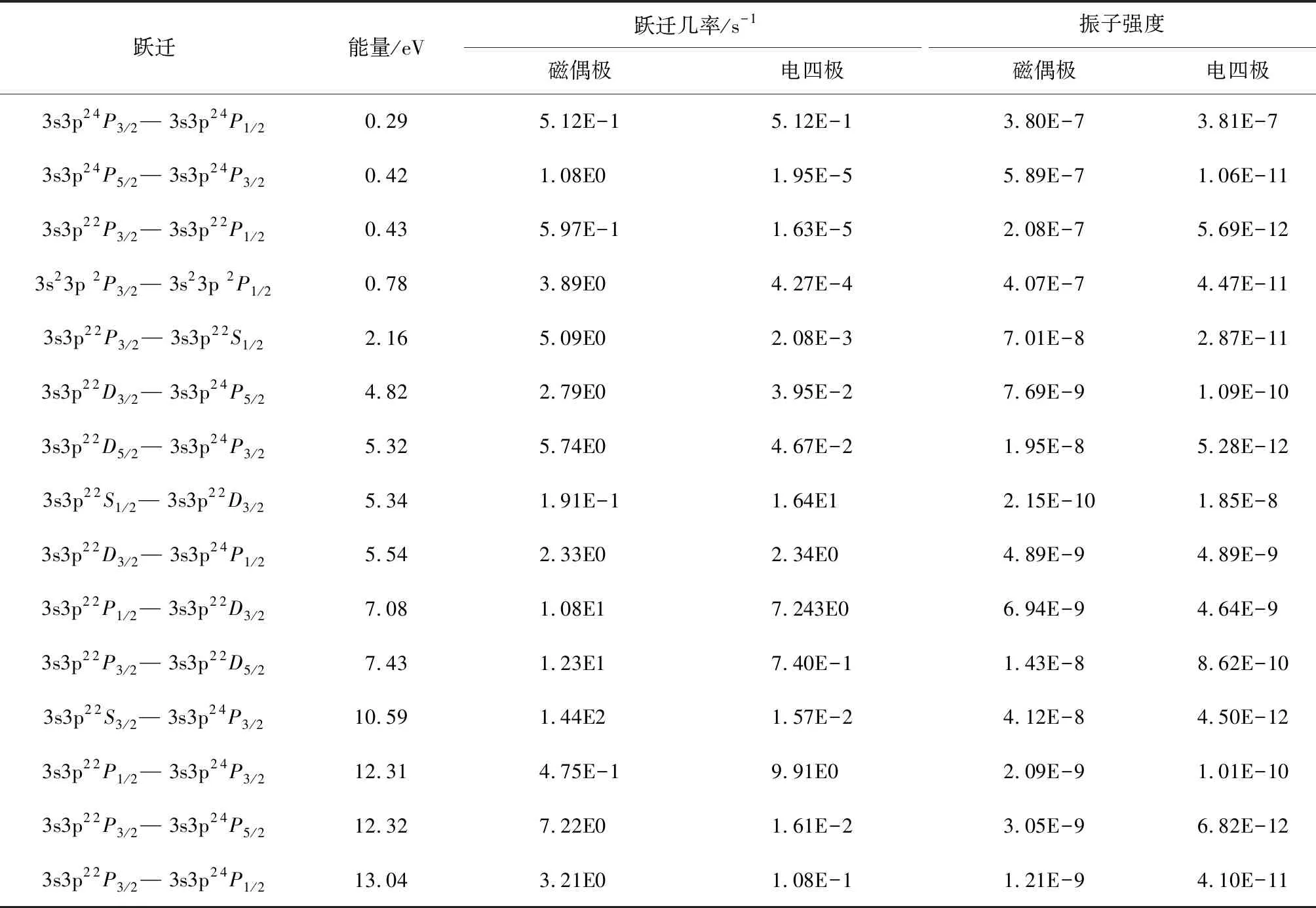
表 5 类铝钛离子3s23p—3s3p2磁偶极与电四极跃迁的能量、跃迁几率与振子强度
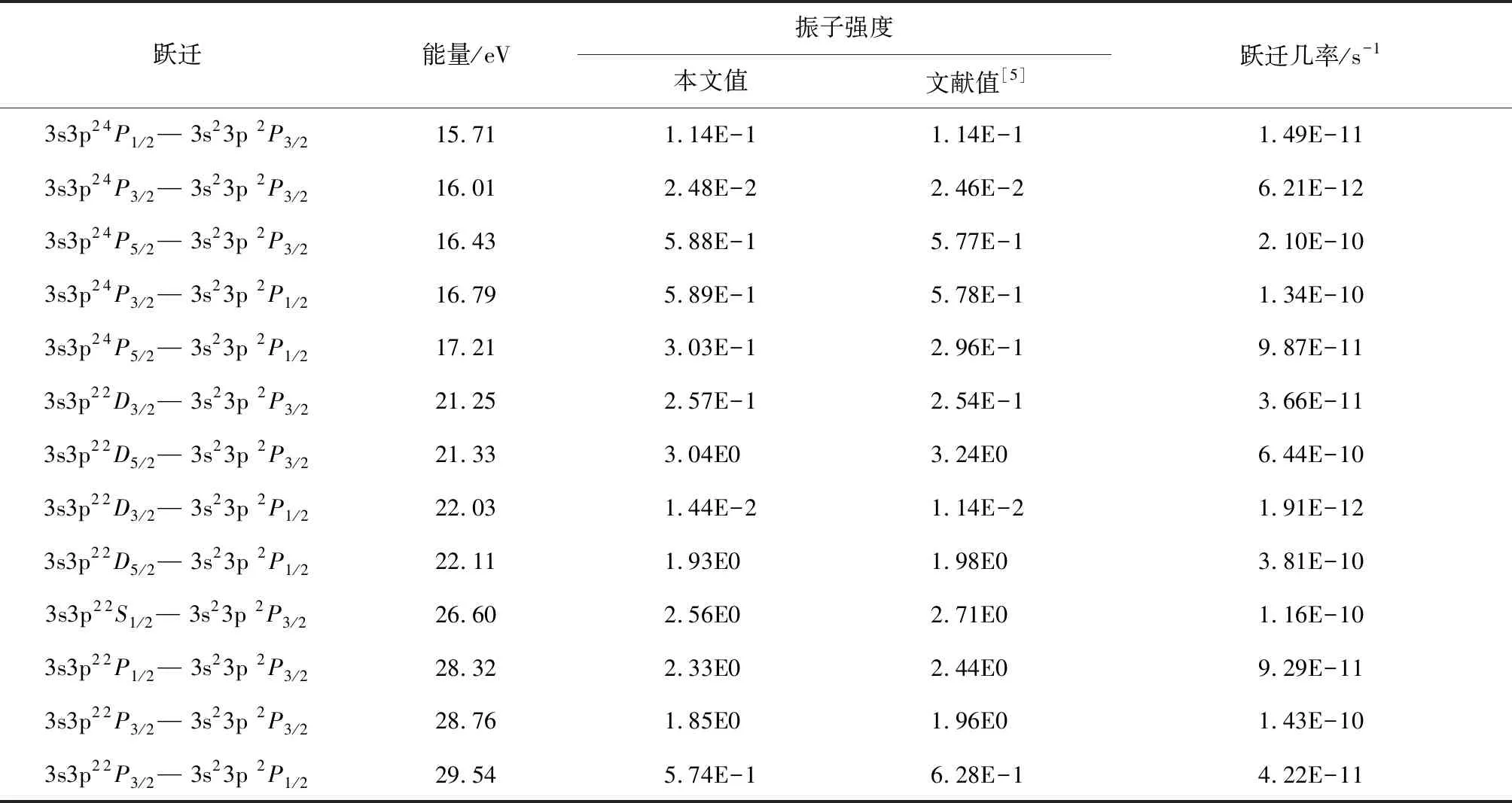
表 6 类铝钛离子3s23p—3s3p2磁四极跃迁的能量、跃迁几率与振子强度
3 结论
利用多组态Dirac-Hartree-Fock方法详细计算了类铝钛离子的3s23p和3s3p2组态,以及两能级之间的电偶极、电四极、磁偶极与磁四极跃迁的波长、跃迁几率和振子强度.结果表明考虑电子关联效应后,当前CV关联效应的结果与已有的实验结果及理论结果符合很好.这些结果对于理解类铝钛离子不同效应有重要的意义,同时对于分析已有的实验结果和指导未来的实验也有重要的意义.需要指出的是当前计算结果是在重点考虑3s23p和3s3p2组态基础上给出的,因此可以看出本文的一些计算结果与实验值仍有一定的差距,因此在今后的计算中,需要考虑更多的组态以获得更好的结果.
