Reduced lysosomal acid lipase activity: A new marker of liver disease severity across the clinical continuum of non-alcoholic fatty liver disease?
2019-08-26FrancescoBarattaDanielePastoriDomenicoFerroGiovannaCarluccioGiuliaTozziFrancescoAngelicoFrancescoVioliMariaDelBen
Francesco Baratta, Daniele Pastori, Domenico Ferro, Giovanna Carluccio, Giulia Tozzi, Francesco Angelico,Francesco Violi, Maria Del Ben
Abstract Lysosomal acid lipase (LAL) plays a key role in intracellular lipid metabolism.Reduced LAL activity promotes increased multi-organ lysosomal cholesterol ester storage, as observed in two recessive autosomal genetic diseases, Wolman disease and Cholesterol ester storage disease. Severe liver steatosis and accelerated liver fibrosis are common features in patients with genetic LAL deficiency. By contrast, few reliable data are available on the modulation of LAL activity in vivo and on the epigenetic and metabolic factors capable of regulating its activity in subjects without homozygous mutations of the Lipase A gene. In the last few years, a less severe and non-genetic reduction of LAL activity was reported in children and adults with non-alcoholic fatty liver disease (NAFLD),suggesting a possible role of LAL reduction in the pathogenesis and progression of the disease. Patients with NAFLD show a significant, progressive reduction of LAL activity from simple steatosis to non-alcoholic steatohepatitis and cryptogenic cirrhosis. Among cirrhosis of different etiologies, those with cryptogenic cirrhosis show the most significant reductions of LAL activity. These findings suggest that the modulation of LAL activity may become a possible new therapeutic target for patients with more advanced forms of NAFLD. Moreover,the measurement of LAL activity may represent a possible new marker of disease severity in this clinical setting.
Key words: Non-alcoholic fatty liver disease; Non-alcoholic steatohepatitis; Lysosomal acid lipase; Cirrhosis; Wolman disease; Cholesterol ester storage disease
INTRODUCTION
Lysosomal acid lipase (LAL) is a key enzyme for intracellular lipid metabolism, which regulates the intra-lysosomal hydrolysis of cholesterol esters (CE) and triglycerides(TG), producing free cholesterol and fatty acids[1]. The LAL activity reduction causes intra-lysosomal accumulation of CE and lowers free cholesterol in cytosol[2]. This reduction increases transcription factor sterol regulatory element binding protein activity, which promotes lipogenesis and synthesis of cholesterol and of very lowdensity lipoproteins. In addition, there is a reduction in liver X receptors expression resulting in impaired cholesterol efflux and high-density lipoprotein (HDL)production[3]. Moreover, low-density lipoprotein (LDL) receptor synthesis and the receptor-mediated LDL uptake are amplified.
In patients with both heterozygous or homozygous deletion of lipase A (LIPA)gene, a lipid phenotype similar to that observed in patients with familial hypercholesterolemia (FH) has been described[4,5]. Therefore, in presence of hypercholesterolemia with type IIa phenotype, it is very important, but not always easy, to make a differential diagnosis with heterozygous FH (Table 1). A family history for early cardiovascular disease and/or hypercholesterolemia supports a diagnosis of heterozygous FH. On the contrary, in the absence of diagnostic criteria for FH, a LAL defect could be suspected, especially in patients with hypercholesterolemia associated with low levels of HDL cholesterol. The Dutch Lipid Clinic Network score[6,7]or the Simon Broome criteria[8]for FH may be two useful tools for a differential diagnosis.
GENETICS OF LAL DEFICIENCY
LAL deficiency (LAL-D) is a rare autosomal recessive genetic disease due to a mutation in the LIPA gene, characterized by the presence of CE and TG in numerous tissues. The most common mutation is the E8SJM variant, which has an estimated frequency of 0.00025 in the general population (i.e., 1 carrier per 200 individuals in Western countries). The LAL-D is a heterogeneous disorder that may present with two different phenotypes based on residual LAL activity levels (Figures 1): Wolman’s disease and cholesterol ester storage disease (CESD)[1,4].
Wolman’s disease starts prematurely during the 6th or 7th mo of life and quickly leads to death, with only a small proportion of patients surviving beyond the first year of age. Infants with LAL-D show delayed growth, associated with signs of malabsorption, hepatosplenomegaly, severe hepatic dysfunction, rapidly progressive anemia, and multi-organ failure; the adrenal calcifications are the pathognomonic sign of Wolman’s disease. In these patients, LAL activity is almost null[1].
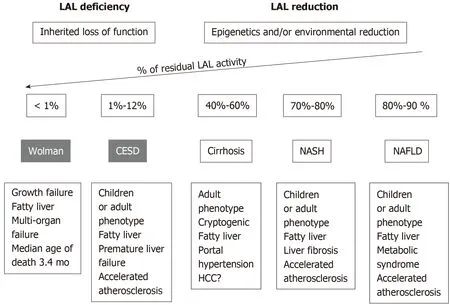
Figure 1 LAL activity reduction in the spectrum of NAFLD. LAL: Lysosomal acid lipase; NAFLD: Non-alcoholic fatty liver disease; NASH: Non-alcoholic steatohepatitis; CESD: Cholesterol esters storage disease.
CESD is the late onset phenotype, being manifested during childhood, adolescence,or in adulthood, with onset age ranging from 5 to 44 years or more. It presents with hepatic steatosis, high levels of aminotransferase, hepatomegaly, and dyslipidemia.As the clinical manifestations of CESD are not very characteristic, the diagnosis is often occasional. The clinical phenotype and disease severity are very variable and depend on the residual enzymatic activity, which is usually less than 12%. Therefore,the coexistence of hepatic steatosis and hypercholesterolemia, in particular in nonobese subjects, should lead to the differential diagnosis between LAL-D and other metabolic causes of non-alcoholic fatty liver disease (NAFLD), such as metabolic syndrome, type II diabetes, hypertriglyceridemia, and central obesity[9].
LIVER ALTERATIONS IN LAL-D
LAL-D leads to CE and TG accumulation in hepatocytes and liver-resident macrophages (Kupffer cells) with subsequent progression to fibrosis (Figures 2). The high prevalence of severe fibrosis in LAL-D and its rapid progression to cirrhosis suggest that lysosomal CE and TG accumulation is a potent driver of liver fibrosis[9-11].A recent study showed increased transaminases in hepatocyte-specific LAL-deficient mice (Liv-Lipa -/-) as well as upregulation of hepatic cytokines and chemokines,known to drive inflammation and leading to Kupffer cell activation and liver damage[12]. In addition, lysosomal CE accumulation induces Kupffer cell activation,causing inflammation and liver damage in high fat/high cholesterol fed Liv-Lipa-/-mice. These findings indicated that hepatocytes’ LAL plays a critical role for liver homeostasis and function.
A recent study reported data on allograft recurrence, liver failure, and other clinical outcomes in 18 liver transplantation (LT) LAL-D patients. LT was necessary for treatment of LAL-D-associated liver failure but, interestingly, did not correct LAL activity, which remained deficient post-LT[13]. Therefore, LT does not correct deficient LAL enzyme in bone marrow derived histiocytes; moreover, LT does not even prevent multi-organ disease progression or liver disease recurrence, as observed in liver biopsies within the first year following LT.
In addition, Burton et al[14]have shown how, in patients with genetic LAL deficiency, 20 wk treatment with enzyme replacement therapy (Sebelipase-alpha) is able to reduce hepatic fat evaluated by magnetic resonance. In addition, treated patients showed serum liver enzymes and serum lipids improvement.
LAL ACTIVITY REDUCTION AND NAFLD
The term NAFLD indicates a set of diseases associated with the presence of excessive accumulation of hepatic fat in the absence of chronic viral infection and alcohol abuse.NAFLD is the most common hepatic disease. It is estimated that the prevalence in the general population is about 20%-30%, reaching up to 70%-90% in the obese or diabetic population[15].
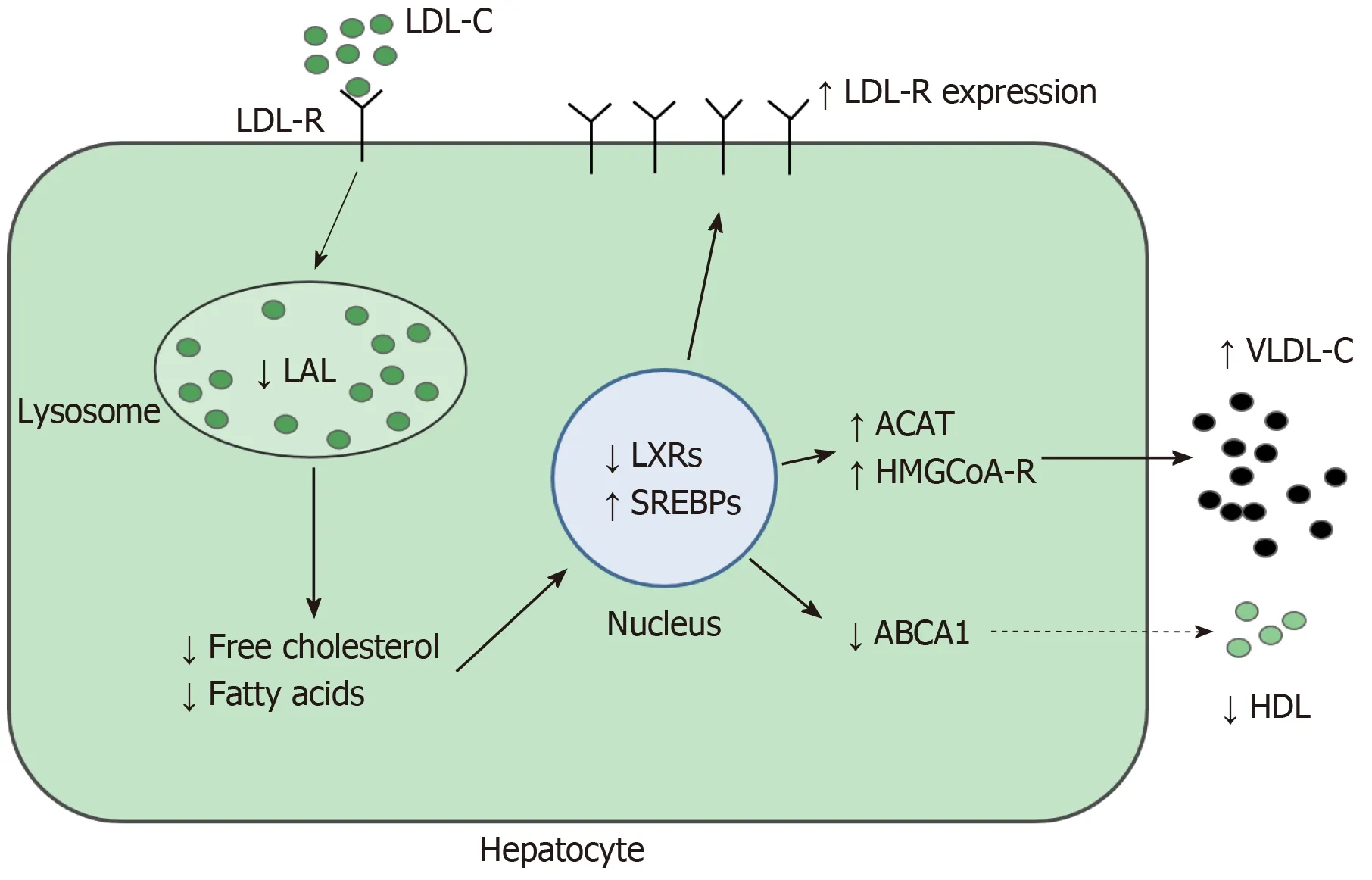
Figure 2 Changes of hepatic lipid metabolism in lysosomal acid lipase deficiency. Reduced lysosomal acid lipase activity causes lysosomal lipid accumulation and reduction of free fatty acids and cholesterol in cytosol. This reduction influences numerous gene transcriptions via transcription factors such as liver X receptors and steroid regulation binding proteins, resulting in higher expression of low-density lipoprotein receptor, acetyl-coenzyme A acetyltransferase, and 3-idrossi-3-metilglutaril-coenzima A reductase and in a lower expression of ATP-binding cassette A1. These changes result in amplified lysosomal lipid accumulation, increased serum very low-density lipoproteins, and decreased serum high-density lipoprotein. LAL: Lysosomal acid lipase; ACAT: Acetyl-coenzyme A acetyltransferase; HMGCoA: 3-Idrossi-3-metilglutaril-coenzima A; LXRs: Liver X receptors; SREBPs: Steroid regulation binding proteins; ABCA1: ATP-binding cassette A1; LDL: Low-density lipoprotein; VLDL: Very low-density lipoproteins; HDL: High-density lipoprotein; LDL-r: Low-density lipoprotein receptor.
NAFLD, in the initial phases, presents as simple steatosis, whose main histological finding is the presence of predominantly macrocytic steatosis in at least 5% of hepatocytes. In some cases, simple steatosis evolves into non-alcoholic steatohepatitis(NASH), in which the histological picture includes steatosis, ballooning, and inflammation with a progressive increase in fibrosis. In the past, NAFLD was considered a benign condition; however, recent evidence suggests a less favorable prognosis, due to the possible evolution in cirrhosis, hepatocellular carcinoma, and hepatic failure[16]. Today, NAFLD is considered the main cause of cryptogenic cirrhosis, the prevalence of which is increasing in recent years, especially in patients with history of obesity and type II diabetes. NAFLD is the second indication for liver transplantation in the US and is expected to exceed hepatitis C virus (HCV) in the next few years, becoming the first cause for liver transplant[17].
Numerous pathogenic factors contribute to the accumulation of lipids in the hepatocytes and, in a proportion of patients, the development of fibrotic processes[18].Among them, insulin resistance, oxidative stress, and low-grade chronic inflammation supported by the production of cytokines deriving from visceral fat. However, the pathogenic mechanisms underlying the progression of simple steatosis to NASH and cirrhosis are not yet fully clarified nor are tools available to predict the evolution of NAFLD.
Prospective studies suggest that the first cause of death in patients diagnosed with NAFLD is cardiovascular disease[19,20]. Atherosclerosis is very common in these subjects, and many of them, especially before the onset of liver complications, develop coronary heart disease[21]. The relationship between NAFLD and cardiovascular risk has long been investigated[15,22-24].
According to the "multiple parallel hits hypothesis", many insults, including insulin resistance, the presence of gene variants, oxidative stress, and alteration of the intestinal microbiota, act simultaneously on the liver causing lipid infiltration and inflammation[18]. It remains to be clarified whether it is the metabolic syndrome that promotes steatosis through insulin resistance, or whether it is NAFLD that induces hyperinsulinemia through a defective mechanism of insulin degradation. The current opinion suggests a bidirectional link between NAFLD and the metabolic syndrome[25].
Few studies have so far assessed the activity of LAL in patients with NAFLD, and the possible role of LAL as one of the multiple hits in NAFLD pathogenesis is under debate[26].
Our group[27]has recently demonstrated reduced LAL activity in patients with NAFLD. LAL activity was significantly reduced in 240 patients with NAFLD compared to 100 heathy subjects (HS) [0.78 (0.61-1.01) nmol/spot per hour vs 1.15(0.94-1.72) nmol/spot per hour, P < 0.001]. An even more marked reduction was observed in patients with histologically diagnosed NASH [0.67 (0.51-0.77) nmol/spot per hour, P < 0.001 vs HS; P < 0.001, between the groups]. In addition, patients with NAFLD who exhibited enzymatic activity below the median had higher serum total cholesterol (P < 0.05) and LDL cholesterol (P < 0.05) and higher levels of transaminases and gamma-glutamyltransferase (alanine aminotransferase, P < 0.001;aspartate aminotransferase, P < 0.01; gamma-glutamyltransferase, P < 0.01).
Therefore, based on our findings, it was possible to hypothesize that the reduction of LAL activity, as well as a predisposing factor for the development of NAFLD, could be considered as a further pathophysiological mechanism for progression to NASH and eventually to cryptogenic cirrhosis. Moreover, based on the above observations[28],we could also hypothesize that LAL activity can constitute a possible tool for the identification of the subjects with more advanced forms of NAFLD and possibly for the monitoring of the response to therapy.
Shortly thereafter[29], a significant reduction in LAL activity in a series of pediatric cases was also observed. In this study, children with significant fibrosis (stage 2-3, n =64) had a significantly lower LAL activity compared to those with mild fibrosis (stage 0-1, n = 104), suggesting a potential role of reduced LAL activity in the pathogenesis of NAFLD-induced fibrosis.
In a further study[30], we found that NAFLD patients disclosed a relatively high prevalence of spleen enlargement and splenomegaly, which were significantly associated with a reduced LAL activity, suggesting that LAL may contribute to spleen enlargement in this setting. Although the degree of LAL activity reduction was less pronounced compared to genetic forms of LAL deficiency, which develop severe fat accumulation in the spleen, a similar mechanism may be hypothesized.
More recently, Tovoli et al[31]performed a study of LAL activity in 81 patients with a diagnosis of NAFLD and 78 matched controls with HCV-related liver disease. LAL activity was significantly reduced in NAFLD compared to that in HCV patients,suggesting that NAFLD is characterized by a specific deficit in LAL activity.
Finally, in a study by Gomaraschi M et al[32], 164 patients with biopsy-proven NAFLD were compared with 60 dyslipidemic patients with similar prevalence of metabolic syndrome and 30 controls. LAL activity on dried blood spot (DBS) was reduced in NAFLD patients compared to controls and those with dyslipidemia.
Therefore, we may conclude that the reduction of LAL activity, even in the absence of genetic diseases, seemed to be associated with the development of progressive hepatic steatosis (Table 2).
LAL-D AND LIVER CIRRHOSIS
Based on follow-up studies, 25% of patients affected by NASH may develop cirrhosis and eventually hepatocellular carcinoma. In fact, current knowledge strongly indicates that cryptogenic cirrhosis is, in truth, the evolution of NASH. However, the mechanisms underlying disease progression remain poorly understood. Following evidence from NAFLD, we hypothesized that epigenetic and/or environmental modulation of LAL activity could be also an unrecognized contributing factor in the progression to cryptogenic cirrhosis.
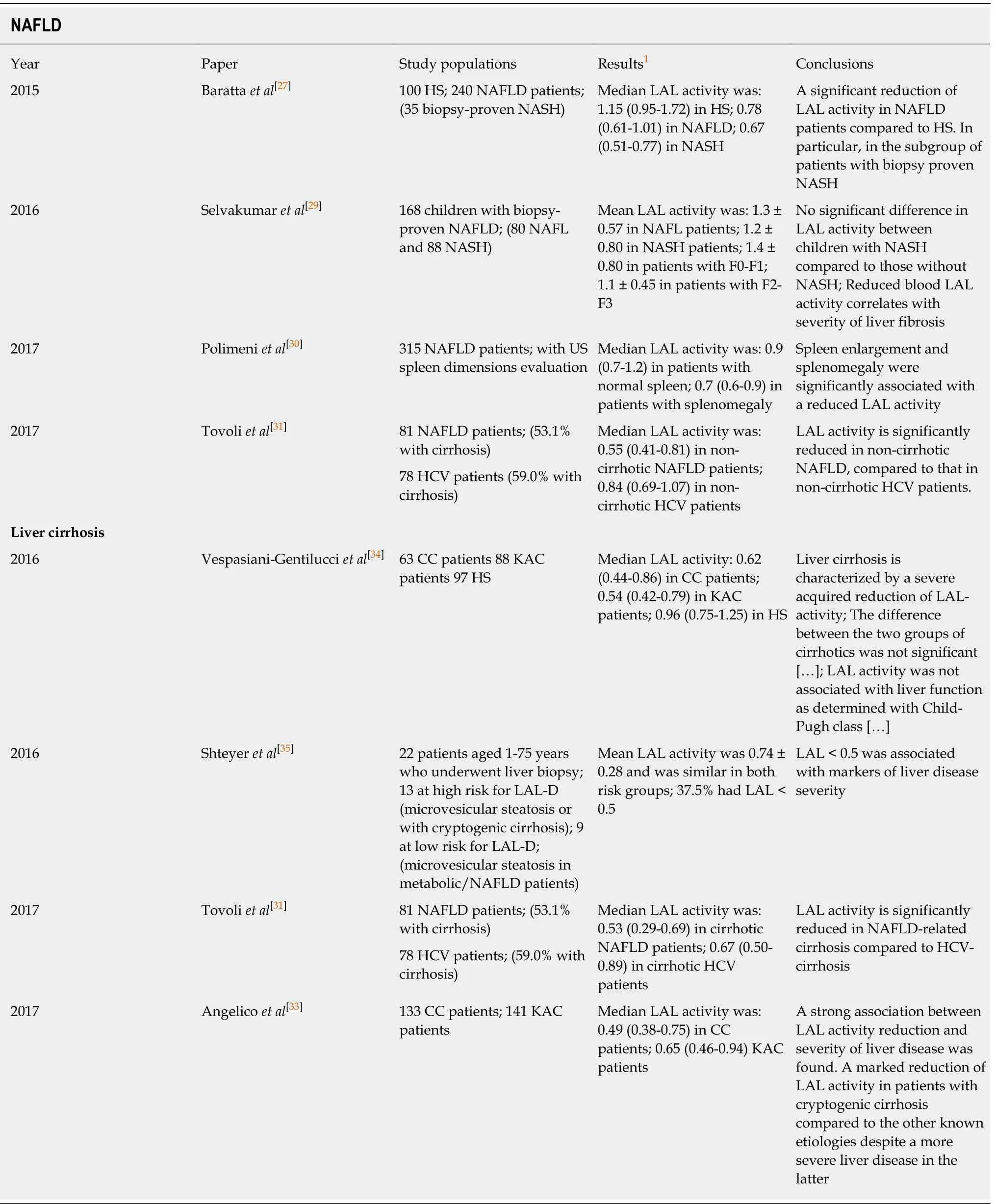
Table 2 Studies investigating the activity of lysosomal acid lipase in patients with non-alcoholic fatty liver disease and liver cirrhosis
To evaluate a possible role of LAL in cryptogenic cirrhosis, we carried out a cohort study including 274 patients with liver cirrhosis of different etiology from 19 centers in Italy[33]. Median LAL activity value was 0.58 nmol/spot per hour, 0.49 and 0.65 in the cryptogenic cirrhosis and alcoholic/viral cirrhosis groups, respectively (P < 0.002).Approximately 30% of patients with cryptogenic cirrhosis showed a severe reduction of LAL activity (i.e., < 0.40 nmol/spot per hour). The reduction was more evident in patients with cryptogenic cirrhosis, despite less severe liver disease. Furthermore, we observed a strong association between LAL activity reduction and severity of liver disease assessed by Child-Turcotte-Pugh and Model for End-Stage Liver Disease scores.
In a further single-center study carried out by Vespasiani-Gentilucci et al[34], LALactivity was severely reduced in patients with cryptogenic cirrhosis with respect to healthy subjects (HS) [0.62 (0.44-0.86) vs 0.96 (0.75-1.25) nmol/spot per hour, P <0.001)], but it was also reduced in known-etiology cirrhotics [0.54 (0.42-0.79)nmol/spot per hour, P < 0.001 vs HS]. In this study, authors sequenced the LIPA gene and excluded genetic etiology for the observed LAL reduction.
Shteyer et al[35]performed a study on medical records of 22 patients with microvescicular steatosis and cryptogenic cirrhosis and 9 with NAFLD diagnosed with liver biopsies. LAL activity inversely predicted liver disease severity, and LAL level of 0.5 was the most sensitive for detecting both histologic and non-invasive markers for disease severity (Table 2).
WHOM TO MEASURE LAL ACTIVITY?
LAL-D should be suspected in non-obese subjects with NAFLD and/or cryptogenic cirrhosis, unexplained persistently elevated levels of transaminases, and/or high levels of LDL cholesterol and low HDL cholesterol.
In these subjects, it would be appropriate to measure LAL activity using the DBS test, which is a simple and inexpensive test that determines the enzymatic activity on the blood spot, subtracting from the activity of the total lipase obtained after addition of a specific LAL inhibitor (Lalistat 2). All patients with a marked reduction in LAL activity (≤ 0.4 nmol/spot per hour) should be investigated for the presence of LIPA gene mutations. Patients homozygous for LAL deficiency have a residual activity equal to or close to 0. The lower limit of the range of normality in the validation tests of the method was 0.8 nmol/spot per hour.
CONCLUSION
A reduced LAL activity may contribute to liver damage in patients with NAFLD, but several issues need to be addressed to better understand the role of LAL in liver diseases. First, there are no reliable in vivo data on LAL activity modulation. In particular, epigenetic and environmental factors that are able to regulate its activity in subjects without homozygous mutations of the LIPA gene have not yet been identified. For example, it is not known whether an intervention on modifiable cardiometabolic risk factors, typically associated with NAFLD, such as metabolic syndrome,diabetes, overweight, or oxidative stress, may affect the modulation of LAL activity in a favorable manner. Moreover, it is still unclear whether low LAL activity may predict liver disease progression and or cardio-metabolic events.
Besides, the specific contribution of circulating cells to the total activity of LAL on the DBS test is still unclear. In a study of a random sample of 300 subjects, LAL activity, measured with 4-methyl-umbelliferyl oleate as the substrate, was present in high concentration in lymphocytes and monocytes but not in polymorphonuclear leukocytes[36]. Recently, in a study performed in 172 HS aged ≥ 18 years, LAL activity in white blood cells was significantly higher than in platelets (458.9 ± 133.6 nmol/mg per hour vs 235.0 ± 88.3 nmol/mg per hour, P < 0.001). However, LAL activity in DBS correlated more strongly with that in platelets, suggesting that platelet count is recommended before interpreting LAL activity in DBS[37].
In addition, it remains to be clarified whether any improvement in the enzymatic activity could result in a reduction of hepatic fat in patients with NAFLD. In light of the results of a recent clinical trial with Sebelipase-alfa, it can be hypothesized that the modulation of LAL activity may become a possible new therapeutic target in the future, even in conditions of less severe reductions of LAL activity[14]. This could mainly concern patients with more advanced forms of NAFLD, such as those with NASH or cryptogenic cirrhosis for which, at present, there are no effective therapies[33,38].
In conclusion, the measurement of LAL activity in patients with NAFLD may be a new non-invasive marker of liver disease severity.
杂志排行

World Journal of Gastroenterology的其它文章
- Exhaled breath analysis in hepatology: State-of-the-art and perspectives
- Miniature gastrointestinal endoscopy: Now and the future
- Issues and controversies in esophageal inlet patch
- Role of hepatocyte nuclear factor 4-alpha in gastrointestinal and liver diseases
- G protein-coupled estrogen receptor in colon function, immune regulation and carcinogenesis
- Helicobacter pylori and cytokine gene variants as predictors of premalignant gastric lesions