Role of hepatocyte nuclear factor 4-alpha in gastrointestinal and liver diseases
2019-08-26MatthewYehDustinBoschSayedDaoud
Matthew M Yeh, Dustin E Bosch, Sayed S Daoud
Abstract Hepatocyte nuclear factor 4-alpha (HNF4α) is a highly conserved member of nuclear receptor superfamily of ligand-dependent transcription factors that is expressed in liver and gastrointestinal organs (pancreas, stomach, and intestine).In liver, HNF4α is best known for its role as a master regulator of liver-specific gene expression and essential for adult and fetal liver function. Dysregulation of HNF4α expression has been associated with many human diseases such as ulcerative colitis, colon cancer, maturity-onset diabetes of the young, liver cirrhosis, and hepatocellular carcinoma. However, the precise role of HNF4α in the etiology of these human pathogenesis is not well understood. Limited information is known about the role of HNF4α isoforms in liver and gastrointestinal disease progression. There is, therefore, a critical need to know how disruption of the expression of these isoforms may impact on disease progression and phenotypes. In this review, we will update our current understanding on the role of HNF4α in human liver and gastrointestinal diseases. We further provide additional information on possible use of HNF4α as a target for potential therapeutic approaches.
Key words: Hepatocyte nuclear factor 4-alpha; Liver cirrhosis; Hepatocellular carcinoma;Viral hepatitis; Gastrointestinal tract; Colorectal carcinoma; Transcription factor
INTRODUCTION
Hepatocyte nuclear factor 4-alpha (HNF4α) is a transcription factor with important roles in liver and gastrointestinal tract development, hepatocyte differentiation, and lipid and glucose metabolism[1]. The HNF4α gene is located on chromosome 20, with transcription regulated by two promoters (P1 and P2) and alternative splicing variants, resulting in 9 distinct isoforms (α1-α9)[1,2]. Adult hepatocytes exclusively express P1 isoforms, while both promoters are active in intestinal epithelia within distinct compartments[1,2]. The importance of HNF4α in development is highlighted by embryonic lethality of gene knockout in mice[3]. Targeted knockout in colonic epithelium disrupts architecture, decreases enterocyte numbers and goblet cell maturation, and perturbs transcriptional profiles[4], while liver-targeted knockout results in hepatomegaly with altered liver architecture, and decreased glycogen storage[5]. Furthermore, expression of HNF4α in mesenchymal stem cells is sufficient to induce epithelioid changes and some hepatocyte functionality including urea production and albumin secretion[6].
The regulation of HNF4α expression, activity, and localization is highly complex(reviewed in[1,7]), reflective of downstream transcri-ptional networks with diverse functional roles including drug metabolism, bile acid synthesis and conjugation, lipid homeostasis, gluconeogenesis, ureagenesis, cell adhesion, proliferation, and apoptosis[7]. Regulation of HNF4α occurs at multiple levels: Epigenetic[8,9]; transcriptional, including promoter regulation, transcript secondary structure, and microRNA-mediated inhibition[7,10]; and post-translational, including protein phosphorylation, degradation, and nuclear localization[1,11,12]. HNF4α transcription factor binding sites are also widely dispersed in the human genome, as evidenced by changes in mRNA levels of ~2500 genes upon over-expression of HNF4α in cell culture[13]. HNF4α expression and activity are altered in numerous disease states involving multiple organ systems, and immunohistochemical detection in the clinical setting has potential diagnostic and prognostic value. This review article updates the understanding of HNF4α role in liver and gastrointestinal pathogenesis and presents potential therapeutic approaches and strategies for possible treatment based on HNF4α involvement.
HNF4ALPHA ACTIVITY IN LIVER PATHOGENESIS
HNF4α perturbations in disease states have been most extensively investigated in the context of liver disease. Most of the major liver diseases have been associated with altered HNF4α expression, isoform ratios, and localization, including inflammation,alcoholic liver disease, non-alcoholic fatty liver disease, fibrosis and cirrhosis, viral hepatitis, and the hepatocellular carcinoma (Table 1). Most commonly, HNF4α expression at the protein and transcript levels is decreased across liver diseases, an observation substantiated by human, animal model, and cell culture-based studies in many cases (Table 2). Altered expression of HNF4α in response to relatively nonspecific stimuli, such as inflammation[14]and injury-induced acute phase response[15],as well as in a remarkable spectrum of disease suggests centrality of HNF4α in response to most modes of hepatocyte injury and stress resulting in de-differetion state of liver function (Table 1, Figure 1)[16]. There is, therefore, a critical need to understand the role of HNF4α in the molecular epidemiology of the de-differentiation state of liver function across these liver diseases for the potential restoration of normal liver function.
Alcoholic liver disease
Expression of HNF4α and carboxylesterase 1 (CES1), an enzyme involved in triglyceride metabolism, was reduced in patients with alcoholic steatohepatitis and mice with methionine and choline-deficient diet-induced inflammation[17]. Alcohol repressed both HNF4α and CES1 expression in primary hepatocyte cell culture.Knockout of CES1 in mice exacerbated alcohol-induced steatosis and steatohepatitis,as well as diet-induced liver inflammation[17].
Non-alcoholic fatty liver disease
HNF4α has important roles in liver lipid and lipoprotein metabolism[18]. Hepatocytetargeted knockout of HNF4α in mice resulted in lipid accumulation, changes in VLDL secretion and bile acid uptake, and alterations in peripheral blood cholesterol and triglycerides[19]. Acute knockout of HNF4α in adult mice resulted in hepatocyte proliferation and vacuolization, and hepatomegaly[20].
HNF4α mRNA and protein levels were decreased in patients with non-alcoholic steatohepatitis (NASH), as well as in cultured hepatocytes and in livers of mice with genetic obesity (ob/ob) or on high fat diet[18]. Network analysis of transcriptomic data in patients with non-alcoholic steatohepatitis identified HNF4α as a central regulator,although transcription of HNF4α itself was not significantly altered[21]. In contrast to a prior study[18], limited immunohistochemistry on 12 liver specimens of patients with NASH, non-alcoholic fatty liver disease (NAFLD) activity (NAS) score 5-7, showed minimal increased immunoreactivity (24%-40% positive cells) compared to a single control (17% positive cells)[21]. Cytoplasmic retention of HNF4α in high fat diet mice with steatosis has also been observed, corresponding to reduced transcription of target genes and HNF4α phosphorylation by protein kinase C isotypes[22]. Taken together, these studies consistently indicate a significant decrease in HNF4α activity in NAFLD, although observations of HNF4α expression levels and localization are less uniform. This could be related to disruption of the transcription factor network responsible for the de-defferentiated state, which is partially controlled by HNF4α activity (Figure 1).
Hepatic fibrosis and cirrhosis
To explore and establish an understanding of the molecular basis for decreased HNF4α activity across US racial population, we performed a mass spectrometrybased proteomics study using clinical tissue samples from Caucasian Americans (CA)and African Americans (AA) and demonstrated, for the first time, that differentially expressed proteins (DEPs) in cirrhotic livers are actually distinct from hepatocellular carcinoma (HCC) and the expression of these proteins are also racially dependent[23]. For example, Figure 2C shows the heat map (truncated) of DEPs between cirrhotic and HCC groups, and that there is a high degree of interaction between HNF4α (a focus hub) and some of these DEPs like serotransferrin (TF) and apolipoprotein lipase A1(APOA1) (Figure 2E). Note also that the level of TF and APOA1 proteins in AA cirrhotic and HCC samples (Figure 2F, upper panel) is equal as in CA protein samples. Furthermore, the levels of HNF4α protein in AA samples are lower compared to CA samples (Figure 2F, lower panel). It is known that AA patients with cirrhotic liver and HCC usually have elevated levels of serum markers of iron stores and altered cholesterol and triglyceride levels[24,25], hence the levels of both are elevated in AA samples. The expression of both TF and APOA1 genes is known to be regulated by the transcription factor HNF4α[26]. The differential expression of HNF4α has been shown in colitis and colitis-associated colon cancer[2], and more recently by our group[27]in cirrhotic livers and HCC. In our study, we used immunohistochemistry to validate the expression of HNF4α isoforms in HCV cirrhotic livers and HCC tissues among CA and AA tissue samples (Figure 3). As shown in Figures 4A and 4B,the staining reactivity of P1- HNF4α isoforms are lower in cirrhotic HCV livers of AAs(grey bars) as compared to CAs (black bars), whereas the observed increase in P1/P2-HNF4α staining (Figure 4A) but not P1 staining (Figure 4B) in HCC for AAs relative to CAs suggests a potential involvement of P2- HNF4α.
Increased extracellular matrix rigidity, as shown in CCl4or 5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) induced fibrosis in rat livers, modulates hepatocyte function and cytoskeletal arrangement in part through inhibition of the HNF4α transcriptional network[28]. HNF4α transcriptional repression in the context of cell culture was prevented by treatment with Rho-dependent kinase (ROCK) inhibitor. In rats with hepatic fibrosis induced by dimethylnitrosamine (DEN) or bile duct ligation, forced hepatic expression of HNF4α decreased fibrosis in improved liver function[29].Similarly, forced re-expression of HNF4α improved functionality in isolated hepatocytes and reversed liver failure in a CCl4-induced rat model[30]. These studies indicate that HNF4α expression is decreased in hepatic fibrosis, and forced expression in this setting appears to promote hepatocyte and liver function.
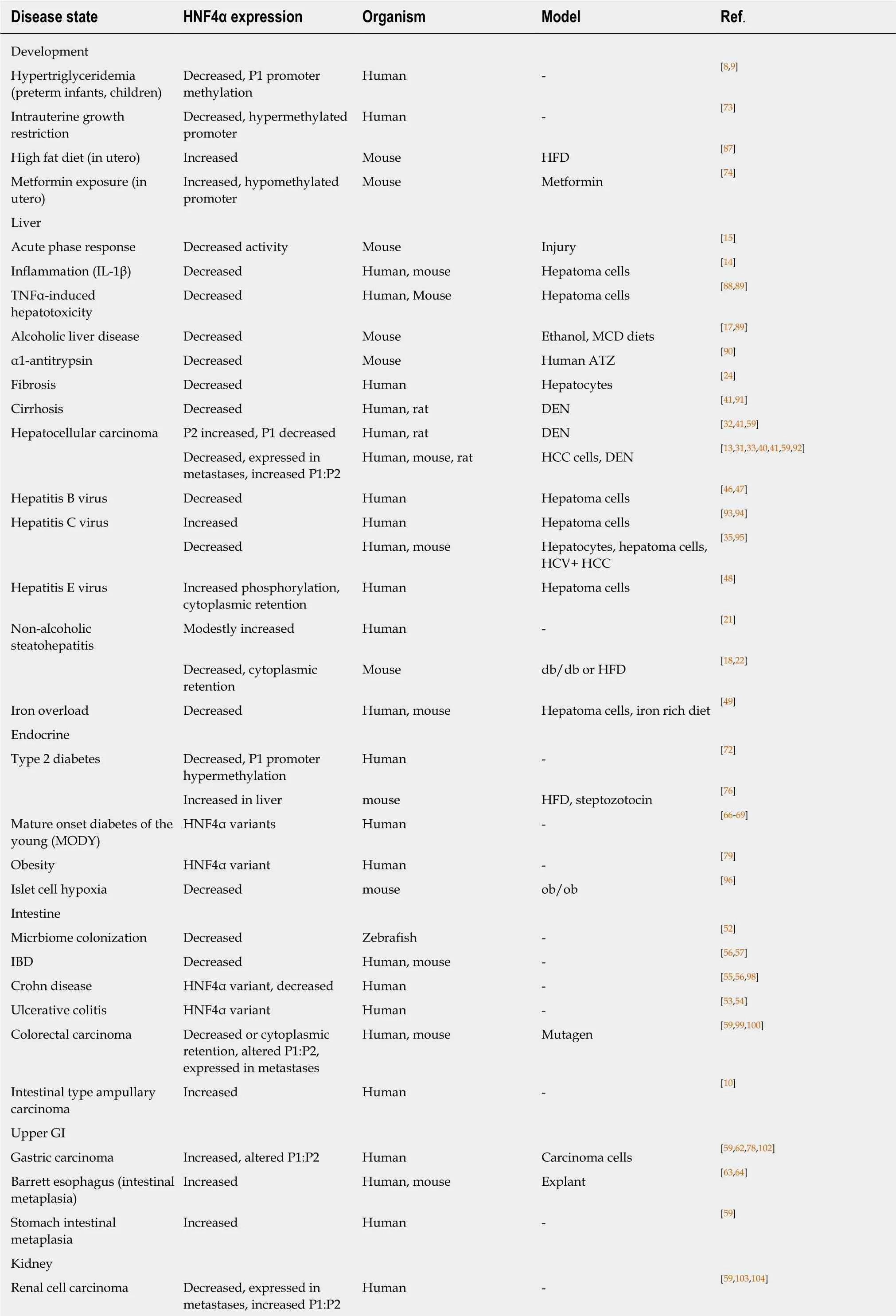
Table 1 Expression of hepatocyte nuclear factor 4alpha and variants in various disease states

IL-1β: Interleukin 1beta; MCD: Methionine-choline deficient; HFD: High fat diet; TNFα: Tumor necrosis factor alpha; ATZ: Mutant Z form of alpha1-antitrypsin deficiency (ATD); HCC: Hepatocellular carcinoma; HCV: Hepatitis C virus; IBD: Inflammatory bowel disease.
Hepatocellular carcinoma
A number of studies have examined HNF4α expression patterns in hepatocellular carcinoma (HCC), including differences in P1 and P2 promoter-derived isoforms.Total HNF4α transcripts were lower in 224 cases of HCC than 220 controls[31]. Rats with DEN-induced hepatocarcinogenesis exhibit decreased hepatic HNF4α expression[29]. However, tissue microarray immunohistochemistry on 615 human HCCs showed inverse correlation of P2 and P1 HNF4α[32]. High P2 HNF4α expression correlated with poor differentiation, vascular invasion, and shorter overall patient survival. Conversely, relatively high P1 HNF4α immunoreactivity in HCC correlated with better differentiation and longer overall survival among a small cohort of 16 patients[33]. However, decreased expression of HNF4α is not uniform in HCC. A series of 196 human HCCs in a heterogeneous background of liver disease, showed 52 (26%)were positive for intense-to-moderate immunoreactivity to HNF4α[34].
HNF4α expression in HCC has been linked to Hippo pathway signaling[31]. Tissue microarray IHC on 75 HCCs revealed increased immunoreactivity for yes-associated protein 1 (YAP1), and lower HNF4α than adjacent tissues[31]. The YAP1/ HNF4α ratio increased with high Edmondson grade. HNF4α appears to be degraded in a proteasome-dependent pathway in the presence of YAP1, and expression of HNF4α in cultured cells or mice with YAP-mediated HCC (mst1/2 conditional mutant)resulted in decreased liver size, transcription of YAP-TEAD target genes, and Ki67 proliferative indices.
Transient knockdown of HNF4α initiates transformation of immortalized hepatocytes through a feedback loop involving miR-24, IL6R, STAT3, miR-124, and miR-629[35,36]. Hepatocytes with knockdown of HNF4α or overexpression of either miR-24 or miR-629 (both HNF4α suppressors) were capable of tumor formation in nude mice[36]. Delivery of miR-124, a transcriptional target of HNF4α, suppressed tumor growth in HCC xenografts and DEN-treated mice. As a corollary in cell culture,knockdown of HNF4α in hepatoma cells also promoted transcription of genes related to the epithelial-mesenchymal transition (EMT) and neoplasia[37,38].
Conversely, overexpression of HNF4α in hepatoma cell lines induced differentiation into hepatocytes and suppressed HCC growth and metastases[39,40]. Forced HNF4α expression in a rat model of DEN-induced liver carcinoma reduced carcinogenesis and decreased EMT[41]. Expression in fibroblasts actually induced a mesenchymal-to-epithelial transition[5].
HNF4α directly interacts with the promoter and induces expression of apoptosis signal-regulating kinase 1 (ASK1)[13]. RT-PCR of human HCC and surrounding nonneoplastic tissue revealed downregulation of HNF4α in 45 of 60 cases (75%) and corresponding ASK1 downregulation in 44 of 50 cases (73%)[13]. Low ASK1 or HNF4α mRNA levels correlated with larger tumor size and advanced stage. Low ASK1 mRNA also correlated with shorter patient survival, in part due to correlation with tumor size, and ASK1 injection directly into xenograft tumors or systemically in mice reduced growth of HCC[13].
Viral hepatitis
The viral hepatitis are associated with decreased HNF4α and/or activity. The hepatitis B (HBV) viral genome contains an HNF4α binding motif in the promoter core, and viral transcription and regulation are dependent on hepatocyte HNF4α[42-45].Interleukin 35 enhanced HBV replication through enhanced HNF4α binding to the core promoter, an effect impaired by promoter mutation or knockdown of HNF4α expression. However, HBV infection or overexpression of vial protein HBx in hepatoma cells reduced HNF4α expression and downstream transcriptional targets[46,47].
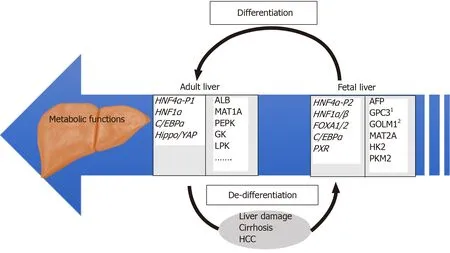
Figure 1 Overview of regulatory and target genes involved in differentiated and de-differentiated stages of liver development. Examples of relevant HNF4α target genes identified by our group[27] are individually numbered.
Electroporation of hepatoma cells with HCV DNA lead to viral replication and decrease in HNF4α mRNA and protein, as well as decreased downstream transcriptional targets[35]. Overexpression of viral proteins including core protein or non-structural proteins (NS5A) was sufficient to significantly decrease HNF4α expression. A transcriptome comparison of hepatocellular carcinomas associated with HCV infection in either African American or Caucasian groups identified differential expression of HNF4α target genes such as SAA1[27]. Immunohistochemistry demonstrated decreased HNF4α expression in HCV positive cirrhosis and hepatocellular carcinoma (n = 72) relative to normal livers, although levels of suppression varied by ethnicity[27].
Hepatitis E virus open reading frame 3 (ORF3) in cultured hepatoma cells resulted in increased HNF4α a phosphorylation, impaired nuclear translocation, and downregulation of target genes[48]. There was no detected effect on HNF4α expression.
Time passed so quickly for both of them that the youth (for now he was quite a young man, and no more a lad) forgot altogether how long he had been there
Iron overload
Iron overload in an iron-rich diet mouse model reduced HNF4α and miR-122 in liver[49]. Liver-targeted adenovirus delivery and overexpression of miR-122 resulted in reduced hepatic inflammation but did not significantly affect iron overload.
HNF4ALPHA ACTIVITY IN COLON PATHOGENESIS
HNF4α plays an important role in colon development[50], and has been implicated in intestinal epithelial differentiation, lipid metabolism, and epithelial junctions[1,51].Expression levels appear to be negatively regulated by gut microbiota, evidenced by a zebrafish model[52]. Altered HNF4α expression and activity, as well as germline variants, have been associated with inflammatory bowel disease (IBD) and colorectal carcinoma[2].
Inflammatory bowel disease
Genome-wide associations studies have linked HNF4α variants with susceptibility to ulcerative colitis in two independent studies[53,54]. An HNF4α P2 promoter single nucleotide polymorphism has also been associated with childhood-onset Crohn disease[55]. In addition to germline variants, HNF4α transcripts were significantly decreased in intestinal biopsies from patients with IBD[56].
Intestine targeted knockout of HNF4α in mice increased susceptibility to dextran sulfate sodium (DSS) induced colitis[1,56]. In another study, knockout of P1 and P2 isoforms of HNF4α in mice resulted in spontaneous intestinal inflammation that worsened with time, leading to epithelial injury, crypt hyperplasia, and proliferation[57]. HNF4α derived from P1 or P2 promoters have distinct effects on colonic epithelium, as demonstrated with an exon swapping mouse model[2]. Mice expressing only P1 promoter-derived α1 isoform HNF4α developed fewer and smaller tumors than wild type mice after treatment with DSS and azoxymethane (AOM), and less susceptibility to DSS-induced colitis. In contrast, expression of only P2 promoterderived α7 isoform HNF4α resulted in greater tumor load and number than wild type mice and were highly sensitive to DSS-induced colitis[2]. HNF4α directly modulated expression of Na+/H+exchanger isoform 3 (NHE3), which has been implicated in IBD pathogenesis[58].
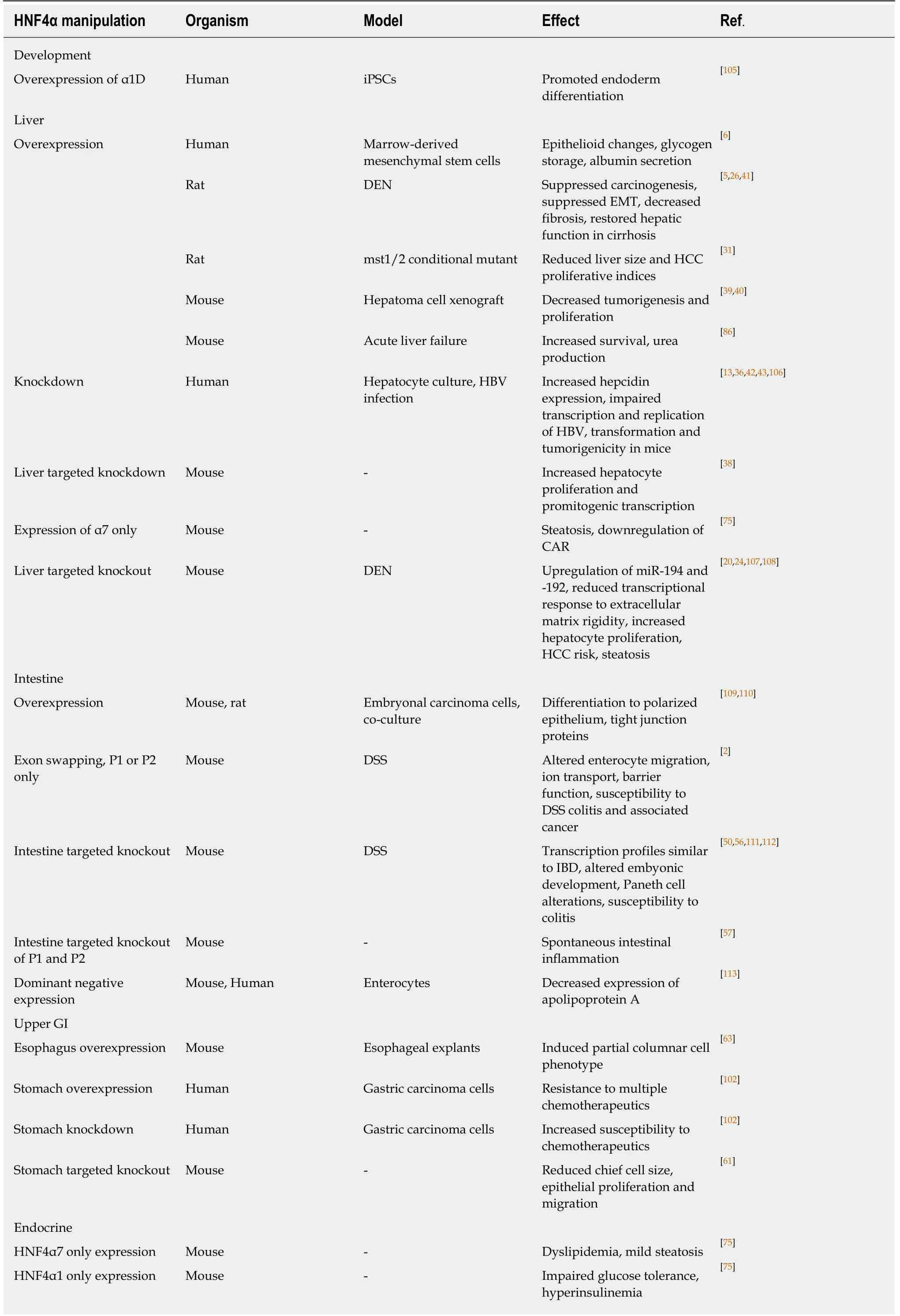
Table 2 Effects of experimental perturbations of hepatocyte nuclear factor 4alpha

iPSCs: Induced pluripotent stem cells; DEN: Diethylnitrosoamine; HBV: Hepatitis B virus; DSS: Dextran sodium sulfate; HNF4α7: Hepatocyte nuclear factor 4alpha7; HNF4α1: Hepatocyte nuclear factor 4alpha1.
Colorectal carcinoma
Isoform-specific HNF4α antibody immunohistochemistry on 18 colorectal carcinomas demonstrated uniform immunoreactivity for P2 and 5/18 (28%) positive for P1[59]; a similar pattern was observed in metastases to lung. Another immunohistochemical study of 450 human colorectal carcinomas revealed either loss or cytoplasmic localization of P1 HNF4α in ~80% of tumors[60]. This pattern appears to be attributable, at least in part, to interaction of HNF4α and Src kinase. Src-mediated phosphorylation of an N-terminal HNF4α tyrosine, present in P1 but not P2 isoforms, influences HNF4α protein stability, transactivation function, and nuclear localization[60]. Consis-tent with HNF4α P1 downregulation being and important feature of colorectal carcinomas,mice expressing only α7 isoform (P2 promoter) HNF4α developed greater tumor load and tumor size than wild type mice in a DSS and azoxymethane (AOM) model[2].Conversely, expression of only the α1 isoform (P1 promoter) resulted in fewer and small tumors than wild type mice.
HNF4ALPHA ACTIVITY IN UPPER GASTROINTESTINAL TRACT PATHOGENESIS
Gastric epithelial development and maintenance are dependent on intact HNF4α.Stomach targeted knockout of HNF4α alters gastric epithelial architecture, with changes including reduced chief cell size and endoplasmic reticulum content,increased proliferation of the stem cell zone, and altered mucous neck cell migration[61].
Transcriptomic analysis of 22 human gastric carcinoma specimens and nonneoplastic controls identified upregulation of HNF4α in carcinoma[62]. P1 promoter HNF4α isoforms were detected in 8 of 14 gastric carcinomas by immunohistochemistry, while normal gastric mucosa had positive immunoreactivity for P2 isoforms only[59]. Knockdown of HNF4α in gastric carcinoma cell lines and xenograft mouse models reduced tumor growth and angiogenesis[62]. Metformin reduced HNF4α expression in gastric carcinoma cell lines and mouse xenografts, and significantly impaired xenograft tumor growth when systemically administered[62].
HNF4α expression appears to be involved in intestinal metaplasia of the upper gastrointestinal tract. Aberrant P1 promoter-driven HNF4α immunoreactivity was observed in gastric intestinal metaplasia, although the number of cases tested is unknown[59]. While HNF4α is not expressed in normal squamous epithelia of the esophagus, HNF4α was expressed along with CDX-2 in esophageal goblet cell metaplasia (Barrett esophagus)[63]. A gene expression profiling study also identified enrichment of HNF4α expression among Barrett esophagus specimens[64].Overexpression of HNF4α in adult mouse esophageal explants resulted in decreased squamous marker such as p63 and induced an expression profile (CK8, E-cadherin,and villin positive) suggestive of a columnar phenotype[13].
HNF4ALPHA ACTIVITY IN PANCREAS AND ENDOCRINE PATHOGENESIS
HNF4α variants have been well described as causing maturity onset diabetes of the young 1 (MODY1), characterized by diminished glucose-stimulated insulin secretion and susceptibility to type 2 diabetes[65-69]. In mouse models of pancreatic β cell HNF4α knockout, there was a similar reduction of glucose stimulation of insulin secretion[65,70,71]. The underlying mechanism is related to endoplasmic reticulum homeostasis in pancreatic islet cells[65].
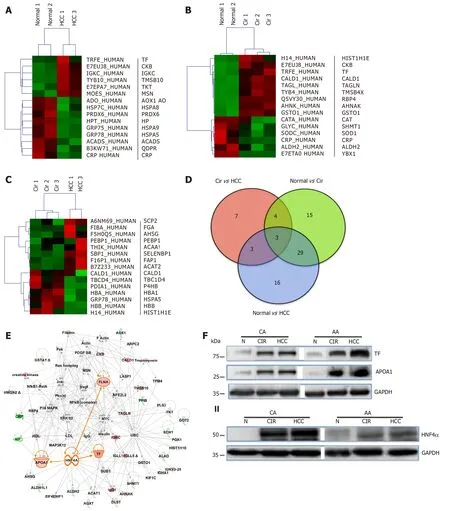
Figure 2 Differentially expressed proteins in normal and liver disease states. Heat maps of differentially expressed proteins (DEPs) (truncated) that were selected following supervised analysis (A) Normal vs. Cirrhosis, (B) Normal vs. Hepatocellular carcinoma, (C) Cirrhosis vs. Hepatocellular carcinoma, and (D) Venn Diagram comparing the significantly DEPs identified. (E) Interactive Network Analysis of DEPs in cirrhosis and hepatocellular carcinoma as compared to normal shows HNF4α as a focus hub to many DEPs. (F) A representative of immunoblot analysis of TF and APOA1 (upper panel), HNF4α (lower panel) in tissue samples of AA &CA. GAPDH was used as a loading control, as published in[23].
Variation in HNF4α expression has also been observed in patients with type 2 diabetes and metabolic syndrome. For example, a monozygotic twin study identified HNF4α P1 promoter hypermethylation as a significant correlate to type 2 diabetes[72]. P1 promoter methylation has also been linked to hypertriglyceridemia in preterm infants and children, as well as intrauterine growth restriction, indicating that epigenetic modulation of HNF4α is linked to the metabolic syndrome[8,9,73]. In contrast,fetuses of metformin-treated mice exhibited hypomethylation of promoter and increased expression of HNF4a[74]. Exon swap mice expressing only HNF4α7 were dyslipidemic with mild hepatic steatosis, and HNF4α1 only expression led to impaired glucose tolerance and hyperinsulinemia[75]. A large genome-wide metaanalysis also identified variants at the HNF4α locus in association with obesity[76].HNF4α and transcriptional target genes were increased in livers of mice with type 2 diabetes in a model of high fat diet followed by streptozotocin injection[76]. In pancreatic cancer cells, HNF4α has been shown to promote resistance to gemcitabine by downregulating hENT1[77]. Therefore, targeting HNF4α might reverse gemcitabine resistance and provide novel treatment strategies for pancreatic adenocarcinoma.
DIAGNOSTIC AND PROGNOSTIC UTILITY OF HNF4ALPHA
HNF4α has been proposed as an immunohistochemical marker useful for pathologic differential diagnosis and prognosis in specific circumstances. HNF4α immunohistochemistry can be helpful in distinguishing gastric primary adenocarcinoma(essentially uniformly positive) from metastatic breast carcinoma (rarely positive)[78-80].A tissue microarray study of 348 lung adenocarcinomas identified 54 cases with positive immunoreactivity for HNF4α, with enrichment among mucinous adenocarcinomas[81]. HNF4α positivity was associated with shorter overall survival among patients with lung adenocarcinoma[81]. However, an independent study of 1021 non-small cell lung carcinomas identified only 20 cases (2% overall, 4% of adenocarcinomas, 11% of mucinous adenocarcinomas) with positive immunoreactivity for HNF4α, and these correlated with absence of lymph node metastases and lower clinical stage[81]. A high percentage of metastatic colorectal carcinomas in lung specimens were positive for HNF4α, but performance of this marker was not a significant improvement over the more commonly used CDX2 and CK20 in this context[81]. HNF4α is also not specific to adenocarcinomas from the GI tract, as HCCs,renal cell carcinomas, and ovarian mucinous neoplasms may be expected to exhibit positive immunoreactivity in some cases[59,80].
Promoter-specific HNF4α antibodies for immunohistochemistry have also revealed substantial variation in neoplastic immunoreactivity for isoforms[59]. For example, 10 of 10 renal cell carcinomas examined were positive for P1 and negative for P2 HNF4α,whereas gastric and colorectal carcinomas were uniformly positive for P2 and variably positive for P1 HNF4α[59].
Hepatocellular carcinomas with β-catenin activation are associated with relatively favorable prognosis, and often exhibit significantly higher uptake of the magnetic resonance contrast agent gadoxetic acid disodium[34]. HNF4α expression correlated with nuclear β-catenin immunoreactivity and expression of the contrast agent transporter OATP1B3, as well as tumor differentiation, indicating potential utility in HCC prognostication.
HNF4ALPHA ACTIVITY AS A THERAPEUTIC POTENTIAL
Given the widespread expression of HNF4α, and demonstrable roles in development,homeostasis, and disease in multiple tissue types, systemically administered direct inhibitors or activators might be expected to exhibit significant undesirable effects. An illustrative example is the opposing effects of HNF4α on fibrosis in cardiac and liver tissue. In cardiac tissue, HNF4α is downstream of AMPK, and upregulation of expression was seen in an angiotensin II-induced cardiac fibrosis mouse model[82].Metformin inhibition or knockout of AMPK reduced cardiac fibrosis, in part by preventing increased HNF4α expression. In contrast, liver fibrosis-associated with decreased HNF4α transcription regulation was restored by forced re-expression of HNF4α which led to reduced fibrosis and reversal fatal liver failure in a rat model[26].These data suggest that de-differentiation state of liver function likely the cause of terminal liver failure and that resetthing the transcription factor network has therapeutic potential for correcting liver failure.
Exposure to flavonoids appears to affect HNF4α expression and activity, although mechanisms underlying these phenomena are unclear. The flavonoid luteolin impaired HBV replication and particle release from cultured HepG2 cells while suppressing HNF4α transcription and reduced viral antigen detection in peripheral blood in a mouse model of acute HBV infection[43]. Treatment of HepG2 cells with the flavonoid derivative 4’-nitro-6-hydroxyflavone reduced expression of the HNF4α target gene microsomal triglyceride transfer protein (MTP) in a transcriptional reporter system[83].
HNF4α antagonists have been described and demonstrated to impair transcription factor activity and exhibit cytotoxicity in human HCC cell lines and xenograft mouse models[84]. The HNF4α antagonist BI6015 also decreased survival of multiple gastric carcinoma cells lines in culture[62]. EC-50 values were estimated in the low micromolar range, but dose-response was non-sigmoidal[62]. The specificity of these compounds, as well as toxicity to non-neoplastic tissues, remains to be fully examined.
Pharmacological manipulation of HNF4α regulatory pathways and transcriptional targets holds promise for therapeutic development. For example, systemic treatment with the AMPK inhibitor metformin impaired gastric carcinoma tumor growth in a xenograft mouse model[62]. Similarly, metformin reduced cardiac fibrosis in an angiotensin II mouse model, an effect correlating to decreased HNF4α expression[82].Systemic administration of the HNF4α transcriptional target miR-124 suppressed hepatocellular carcinoma growth in xenograft and DEN-treated mouse models[36].
Engineered cellular therapies with manipulation of HNF4α have been explored in few studies. Conditioned media of mesenchymal stem cells (MSCs) stably expressing HNF4α inhibited proliferation of SK-Hep-1 and HepG2 cells in culture, and intravenous injection of HNF4α-expressing MSCs into nude mice xenograft models reduced tumor size[85]. Peritoneal injection of immortalized hepatocytes overexpressing HNF4α improved survival and serologic liver enzyme markers in a Dgalactosamine rat model of acute liver failure, as compared to immortalized hepatocyte controls[86].
CONCLUSION
HNF4α is a highly conserved member of nuclear receptors superfamily of liganddependent transcription factors that is expressed in liver, stomach, intestine, pancreas and kidney. HNF4α is known for its role in the liver where it is a master regulator of liver-specific gene expression and essential for adult and fetal liver function (Figure 1). Dysregulation of HNF4α transcriptional activity is linked to several pathological disorders, such as liver cirrhosis, hepatocellular carcinoma, Maturity Onset Diabetes of the Young 1 (MODY1), colitis and colon cancer. Although there are growing evidences for the role of different HNF4α isoforms in the pathogenesis of these diseases, the exact molecular epidemiology and the molecular mechanisms involved are yet to be established. It is anticipated that the identification of specific interacting partners associate with these isoforms in each disease state is essential for differential expression of target genes, and hence signaling pathways. In turns, these targets could be used as diagnostic tools and for the treatment of diseases liked to transcriptional dysregulation of HNF4α.
杂志排行

World Journal of Gastroenterology的其它文章
- Exhaled breath analysis in hepatology: State-of-the-art and perspectives
- Miniature gastrointestinal endoscopy: Now and the future
- Issues and controversies in esophageal inlet patch
- G protein-coupled estrogen receptor in colon function, immune regulation and carcinogenesis
- Helicobacter pylori and cytokine gene variants as predictors of premalignant gastric lesions
- Intestinal enteroids/organoids: A novel platform for drug discovery in inflammatory bowel diseases