Intestinal enteroids/organoids: A novel platform for drug discovery in inflammatory bowel diseases
2019-08-26JunHwanYooMarkDonowitz
Jun-Hwan Yoo, Mark Donowitz
Abstract The introduction of biologics such as anti-tumor necrosis factor (TNF)monoclonal antibodies followed by anti-integrins has dramatically changed the therapeutic paradigm of inflammatory bowel diseases (IBD). Furthermore, a newly developed anti-p40 subunit of interleukin (IL)-12 and IL-23 (ustekinumab)has been recently approved in the United States for patients with moderate to severe Crohn’s disease who have failed treatment with anti-TNFs. However,these immunosuppressive therapeutics which focus on anti-inflammatory mechanisms or immune cells still fail to achieve long-term remission in a significant percentage of patients. This strongly underlines the need to identify novel treatment targets beyond immune suppression to treat IBD. Recent studies have revealed the critical role of intestinal epithelial cells (IECs) in the pathogenesis of IBD. Physical, biochemical and immunologic driven barrier dysfunctions of epithelial cells contribute to the development of IBD. In addition,the recent establishment of adult stem cell-derived intestinal enteroid/organoid culture technology has allowed an exciting opportunity to study human IECs comprising all normal epithelial cells. This long-term epithelial culture model can be generated from endoscopic biopsies or surgical resections and recapitulates the tissue of origin, representing a promising platform for novel drug discovery in IBD. This review describes the advantages of intestinal enteroids/organoids as a research tool for intestinal diseases, introduces studies with these models in IBD, and gives a description of the current status of therapeutic approaches in IBD. Finally, we provide an overview of the current endeavors to identify a novel drug target for IBD therapy based on studies with human enteroids/organoids and describe the challenges in using enteroids/organoids as an IBD model.
Key words: Enteroids; Organoids; Inflammatory bowel diseases; Crohn’s disease;Ulcerative colitis

INTRODUCTION
Inflammatory bowel diseases (IBD) are chronic relapsing-remitting disorders with unknown etiology comprising Crohn’s disease (CD) and ulcerative colitis (UC). IBD has been thought to result from a dysregulated, inappropriate immune response to commensal microbes in a genetically susceptible host with environmental factors(dietary factors, smoking, drugs, etc.) contributing to the disease risk[1,2].
Over the last decades, anti-tumor necrosis factor (TNF) monoclonal antibodies as well as long relied upon conventional therapies (5-Aminosalicylates, corticosteroids,immunosuppressive agents) have been used to induce remission by targeting immune cells or inflammatory mechanisms. Recently developed biologics (anti-integrins, antip40 subunit of IL-12 and IL-23) also act through immunomodulation. These agents have dramatically contributed to increase the rate of clinical remission. Despite advances of biologics, currently, one-third of patients with CD and one sixth of patients with UC require surgery within 5 years after diagnosis[3-8]. This suggests that current anti-inflammatory or immune suppressive strategy is not enough to overcome the heterogeneous, multifactorial IBD. Therefore, new therapeutic strategy targeting other types of cells or mechanisms are needed.
Growing evidence suggests that intestinal epithelial cell (IEC) dysfunction plays a major role in the pathogenesis and perpetuation of IBD[1,9]. Several genetic alterations of IBD are associated with epithelial functions including integrity maintenance(PTPN2, CDH1, PTGER4), secretory defense (XBP1, ATG16L1), and bacterial sensing(CARD15)[9-12]. Mucosal healing achieved by regeneration of IECs is associated with more favorable prognoses in IBD such as decreased relapse, low risk of admission and surgery, and long-term clinical remission[1,9,13]. Several studies have shown that intestinal epithelium of IBD can harbor persistent alterations in gene expression or DNA methylation despite complete endoscopic and histologic remission, which could contribute to disease relapse or perpetuation[9,14-17]. However, no drugs directly targeting changes in IECs in IBD have been developed. This might be attributed to the inability to maintain long-term primary IEC cultures as well as to the previous drug development based on immortalized cell lines or animal models which are mechanistically different from human IBD[18-20].
Recently, remarkable progress has been made in long-term primary IEC cultures called human intestinal enteroids (small intestine) and colonoids (colon)/organoids which develop from human adult intestinal stem cells (ISCs)[21-23]. Ex vivo human enteroid/organoid cultures have diverse advantages over traditional animal models and cell lines in context of recapitulating human in vivo physiology, even after many generations, apparently with limited genetic or physiologic alterations[24].Additionally, intestinal enteroids/organoids can be easily established from endoscopic biopsies in IBD patients and maintain the location or some disease specific features[14,25-28]. Therefore, the intestinal enteroid/organoid culture system represents a promising tool for IBD modeling and drug development focusing on IEC dysfunction.However, the current limitation of this model is that it is not yet known if this model maintains the inflammatory phenotype and epigenetic stem cell modifications that occur in the IBDs.
INTESTINAL ENTEROIDS/ORGANOIDS DERIVED FROM ADULT ISCS
Human “mini-intestines” are derived either from adult ISCs (enteroids/organoids)[23,29]or from induced pluripotent stem cells (iPSCs)(organoids)[30]. The iPSCs-derived intestinal organoids contain both epithelium and mesenchyme including myofibroblasts, smooth muscle cells[29-31]but have limitations of requiring meticulous maintenance and initially mimicking fetal tissue. In contrast, the adult ISCs-derived intestinal enteroids/organoids can be easily established from human tissue (intestinal crypts), making it a tool more accessible to general researchers[29].Thus, this review specifically focuses on intestinal enteroids/organoids derived from an adult ISC origin.
Intestinal enteroids/organoids can be generated from single Lgr5+(Leucine-rich repeat-containing G protein-coupled receptor 5) ISC plus Paneth cells or from intestinal crypts containing ISCs[21-23]. Intestinal crypts can be isolated from surgical resections or endoscopic biopsies, embedded in Matrigel (an extracellular matrixcontaining substance), and cultured as three-dimensional (3D) spheroids in several growth factors (Wnt3A, R-spondin, Noggin, and EGF) enriched media[32]. After withdrawal of critical growth factors, intestinal enteroids/organoids differentiate to mimic IECs in villi composed of mature enterocytes, enteroendocrine cells, goblet cells, and tuft cells while ISCs and transit-amplifying cells are lost[32].
THE ADVANTAGES OF INTESTINAL ENTEROIDS/ORGANOIDS AS A RESEARCH TOOL FOR INTESTINAL DISEASES
Intestinal enteroid/organoid culture system can overcome the limitations of immortalized epithelial cell lines, human fetal intestinal organ cultures, and animal models. In contrast to cell lines which are genetically transformed and thus represent altered genotypes and phenotypes significantly different from those of primary cells[19], the intestinal enteroid/organoid culture is a primary culture system which maintains in vivo characteristics of human intestinal epithelium even after many passages[21]. Furthermore, the current human cancer derived intestinal epithelial cell lines, as normally grown, consist of a single cell type (e.g., Caco-2, HT29: enterocyte[33,34]) and thus fail to recapitulate the diversity of cell types in the normal intestinal epithelium. In contrast, intestinal enteroids/organoids can give rise to all the types of epithelial cells in crypt or villus structure (ISCs, Paneth cells, transit amplifying cells, enteroendocrine cells, goblet cells, enterocytes, tuft cells, and microfold cells)[21,35-37]. Thus, intestinal enteroids/organoids can more closely reflect normal physiology or disease pathogenesis of intestinal epithelium where each single type of IECs interact with other types of IECs by paracrine and autocrine mechanisms[38].
Human fetal intestinal organ culture, which is prepared from intestinal tissue obtained from therapeutic abortions, has been used to study the fetal intestinal immune response to luminal microbes[39]and the pathogenesis of necrotizing enterocolitis[40]and celiac disease[41]. Human fetal intestinal organ culture has a strength of providing multiple cell types and sequential differentiation which are not preserved in intestinal enteroid/organoid cultures. However, in comparison to enteroids/organoids, intestinal human fetal intestinal organ culture has several weaknesses. These include the application of high throughput drug screening,difficulty in obtaining fetal tissue, short viability (up to 48 h), limitation in delivery of exogenous stimuli and real-time monitoring due to the thickness of the intestinal tissue[42].
In the drug development process, animal models have provided a wide range of systemic response, however, these models are resource-intensive (cost, time, labor)and low-throughput[43]. More importantly, the mechanistic differences between animal models and human diseases might be one of the critical factors contributing to drug failure in clinical trials[38]. Meanwhile, intestinal enteroids/organoids-based drug screening can be high throughput by an in vitro culture system, mechanistically similar to human diseases, and thus potentially more precisely predicting drug response in humans. In particular, growing enteroids as polarized monolayers instead of spheroids allows direct apical and basolateral access by pathogens and oral drugs,and subsequently enables the effective study of ion transport and secretory functions.A recent study demonstrated the successful use of enteroid monolayers in drug discovery by miniaturizing mouse colonoid monolayer cultures to 96-well plates, and conducting a phenotypic screen of approximately 2000 drug candidates[44]. We have adopted the following approach for development of anti-diarrheal drugs.Identification of drug targets includes studies in diarrheal models in human enteroid monolayers. Initial drug candidates are screened early for toxicity in human enteroids with further development curtailed if human intestinal toxicity is identified. Once pharmacokinetic approaches are carried out in mouse intestine and human colon cancer cell lines, human enteroids are studied to determine IC50 and if similar it is considered that the specific drug can be further developed. This approach was used with the CFTR inhibitor BPO-27 which is now under development by pharma for phase I and II studies[45].
Human enteroids are amenable to lipofectamine-, low voltage electroporation-, and viral-based genetic manipulation including knock down, knock-out, knock-in, or overexpression[38]. The CRISPR/Cas9 system was also used to edit the genome of intestinal enteroids derived from cystic fibrosis patients and repaired the cystic fibrosis transmembrane conductance regulator (CFTR) function[46].
The indefinitely propagating intestinal enteroid/organoid cultures can be very useful to investigate long-term pathogenesis of intestinal diseases such as chronic inflammation, fibrosis and colitis associated cancer (CAC)[47,48]. In contrast to iPSCsderived intestinal organoids which can acquire genetic and epigenetic variations during culturing[49]or isolated primary IECs which rapidly enter into anoikis(detachment induced apoptosis), ISCs-derived intestinal enteroids/organoids appear to be genetically, epigenetically, and phenotypically stable during long-term culture[9,21,46,50], although more detailed characterization is needed.
Intestinal enteroids/organoids faithfully retain the physiological and pathological features from intestinal segment of origin[25,27]. Previous studies showed that intestinal enteroids/organoids retain the tissue specific (region-specific, age-specific, and disease-specific) transcriptional and epigenetic profiles which might be programmed within ISCs. The long-term cultures of both human and mouse intestinal enteroids/organoids demonstrated that region-specific gene expression(duodenum/jejunum/ileum) is intrinsically imprinted in ISCs, for example, Gata4 in duodenum and jejunum suppresses expression of ileal genes, and that their differentiation fate is independent of region-specific extracellular signals[25]. Another study identified gut-segment specific DNA methylation patterns in human intestinal enteroids/organoids derived from small and large bowel[26]. The DNA methylation patterns were cellular environment-independent and retained in intestinal enteroids/organoids over long-term culture. Interestingly, fetal gut-derived intestinal enteroids/organoids showed marked dynamic changes of DNA methylation and gene expression indicating in vitro maturation. This result suggests that intestinal enteroids/organoids can retain age or development-specific signatures in ISCs[26].Compared with non-IBD subjects, intestinal enteroids/organoids generated from UC patients showed different gene expression profiles (genes associated with antimicrobial defense, secretory and absorptive functions) which are possibly due to permanent changes in ISCs[14]. Moreover, ISCs derived from active CD patients revealed a distinct gene expression pattern of ISC markers (LGR5 and SMOC2)[27]. In a recent study, Howell et al. identified that IECs from children with CD or UC showed specific changes in DNA methylation and transcription which were stable over time and were partly retained in intestinal enteroid/organoid cultures[28]. Table 1 compares the transcriptional or epigenetic features of intestinal enteroids/organoids derived from each intestinal segment or from normal or diseased intestine.
EPITHELIAL BARRIER DYSFUNCTIONS: A PRESSING NEED FOR INTESTINAL ENTEROIDS/ORGANOIDS-BASED IBD RESEARCH
IBD is known to develop from an uncontrolled host immune response to commensal bacteria, with genetic susceptibility and environmental factors[2]. In addition,emerging evidence has revealed the role of IECs as regulators of mucosal immune homeostasis and highlighted the importance of the epithelial barrier function in pathophysiology of IBD, further supporting a pressing need for intestinal enteroids/organoids-based IBD research[1,51]. Intestinal epithelium apically contacts with luminal microbial flora and environmental factors, and basolaterally interacts with lamina propria immune cells. Moreover, several susceptibility genes of IBD are associated with epithelial innate immune functions and barrier functions[2,12]. Thus,IECs are located at the crossroad linking major IBD risk factors. Diverse barrier dysfunctions of IECs which play a key role in IBD pathogenesis are identified as follows.
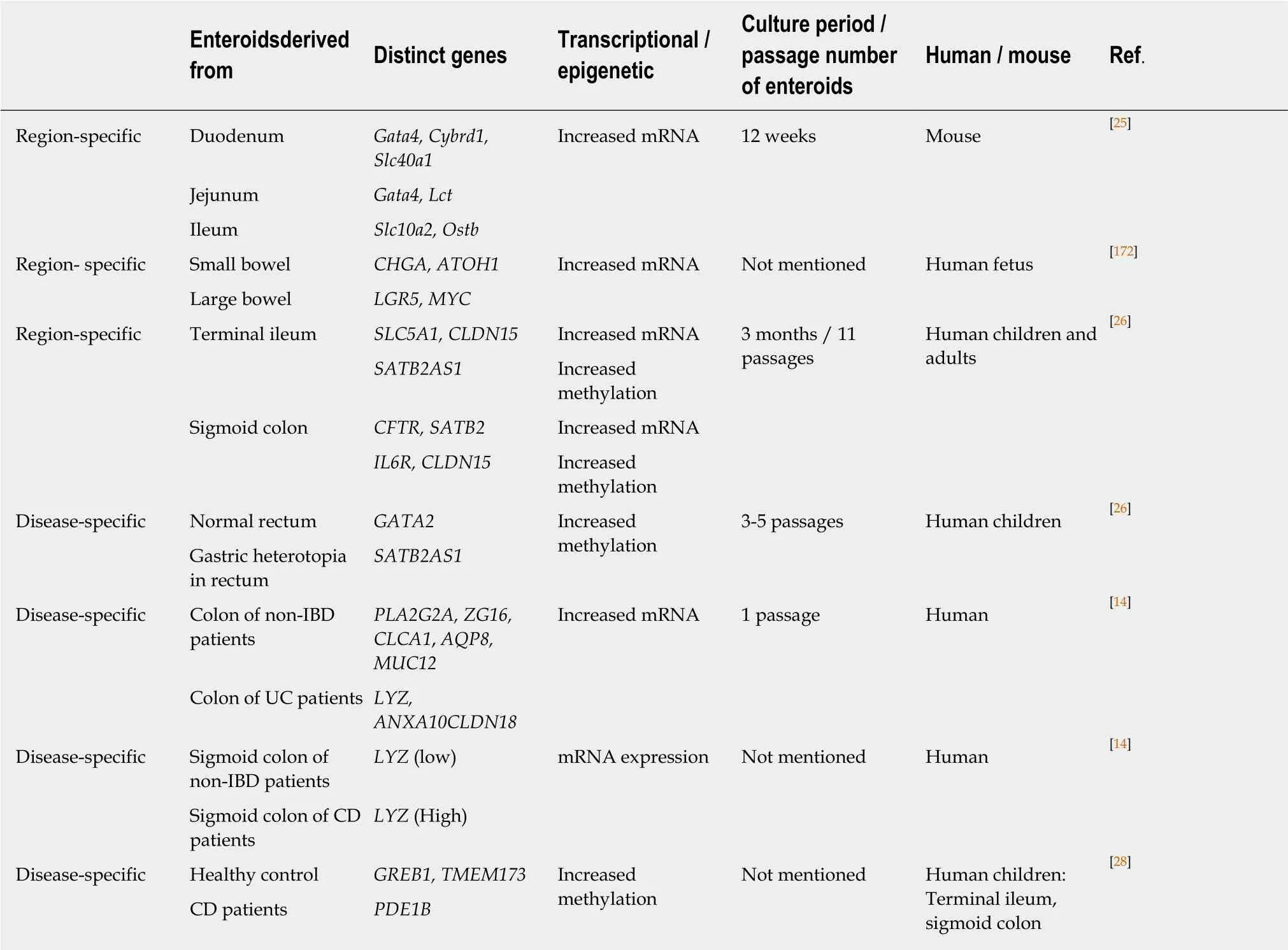
Table 1 Enteroids/organoids retain the tissue specific transcriptional and epigenetic profiles
Gata4: GATA binding protein 4, transcription factor specific to duodenum and jejunum; Cybrd1: Cytochrome b reductase 1 specific for iron uptake; Slc40a1:Solute carrier family 40 member 1, Fe2+transporter ferroportin for iron uptake; Lct: Lactase-phlorizin hydrolase for dissacharide digestion; Slc10a2: Solute carrier family 10 member 2, apical sodium-dependent bile acid transporter; Ostb: Organic solute transporter beta for bile acid uptake; CHGA:Chromogranin A, a marker of enteroendocrine cells; ATOH1: Atonal BHLH Transcription Factor 1, key regulator of the Notch pathway; LGR5: Leucine rich repeat containing G protein-coupled receptor 5; MYC: MYC Proto-Oncogene, BHLH Transcription Factor; SLC5A1: Solute carrier family 5 member 1;CLDN15: Claudin 15; SATB2AS1: Special AT-rich sequence-binding protein 2 antisense 1; CFTR: Cystic fibrosis transmembrane conductance regulator;SATB2: Special AT-rich sequence-binding protein 2; IL6R: Interleukin-6 receptor; GATA2: GATA binding protein 2; PLA2G2A: Phospholipase A2 Group IIA, antibacterial enzyme; ZG16: Zymogen Granule Protein 16, mucus secreting cell marker; CLCA1: Chloride Channel Accessory 1, mucus secreting cell marker; AQP8: Aquaporin-8, water transporter; MUC12: Transmembrane mucin 12; LYZ: Lysozyme C; ANXA10: Annexin-10; CLDN18: Claudin 18; GREB1:Growth regulation by estrogen in breast cancer 1; TMEM173: Transmembrane protein 173; PDE1B: Phosphodiesterase 1B.
Physical barrier dysfunction of IECs
As a physical barrier, IECs form a semi-permeable lining and segregate luminal microbial flora from host immune system to prevent infection or inflammation. IECs maintain physical barrier function by tight junctions (TJs), adherens junctions (AJs),and desmosomes which connect adjacent IECs and regulate intestinal permeability. In IBD, pro-inflammatory cytokines (e.g., TNF-α, IFN-γ) disrupt TJs and increase permeability by cytoskeletal reorganization of IECs[11]. Also, polymorphisms in several IBD susceptibility genes which regulate expression and assembly of TJs and AJs,result in downregulation of junctional proteins leading to increased permeability,facilitating exposure of luminal bacteria and antigens to the host immune system[12].To maintain the continuity of epithelial barrier after tissue injury, the voids created during the extrusion of either apoptotic or necrotic cells are rapidly covered by adjacent cells (termed restitution), followed by more delayed process of wound healing including the reinforcement of freshly developed IECs which are continuously differentiated and proliferated from ISCs[52]. Trefoil factors (TFFs), which are secreted from goblet cells, are known to be the initiators of mucosal healing by promoting restitution and by suppressing IEC apoptosis[53]. Down regulated TFFs in goblet cells have been implicated in the pathogenesis of IBD[54-56](Figure 1A).
Biochemical barrier dysfunction of IECs
Furthermore, as a biochemical barrier, IECs secrete several anti-microbial peptides,secretory IgA and mucin2 (MUC2). Thick mucus layer act as a chemical boundary,preventing pathogenic bacterial invasion by trapping microbes with its viscosity[51]. In active UC, mucus layer is thinner and MUC2 expression is reduced by goblet cell depletion[57]. Recent studies showed the down-regulation of bicarbonate transporter(BEST2, bestrophin2) expression in goblet cells of UC patients[58,59], which is associated with mucus dysfunction and UC pathogenesis. Anti-microbial peptides secreted from Paneth cells (defensins, cathelicidins, lysozyme) or enterocytes (REGIIIγ, regenerating islet-derived protein IIIγ) maintain host immunity by formation of micropores in the bacterial membrane which break bacterial integrity[11,51]. However, in IBD, several genetic defects in autophagy and the unfolded protein response (UPR) result in endoplasmic reticulum (ER) stress and apoptosis, which induce secretory dysfunctions of Paneth cells and goblet cells[11,60-63](Figure 1B). In particular, Paneth cell secretory dysfunction has been implicated in the pathogenesis of ileal CD[60,61,63,64]and the expression of Paneth cell α-defensin has been shown to be decreased in patients with ileal CD[65]. Several genetic susceptibility factors of CD have been linked to Paneth cell dysfunction. These susceptibility factors encode proteins which are associated with pathogen recognition (NOD2)[66], ER stress (XBP1, X-box-binding protein 1), autophagy (ATG16L1, Autophagy-related16 like 1[62]), and defensin production (TCF4, T cell-specific transcription factor 4)[60,61,63,65].
Innate immune dysfunction of IECs
As an immune homeostasis regulator, IECs have well evolved and diverse functions,such as microbial recognition, sampling and transcellular transport of luminal contents (microfold cell), and regulated innate and adaptive immune cell functions[11,51]. In normal physiology, IECs recognize commensal bacteria with pattern recognition receptors (PRR: Toll-like receptor, TLR, NOD-like receptor, NLR) and maintain the mononuclear phagocytes in a tolerogenic phenotype by hyporesponsive PRR signaling[67,68]. Under active IBD conditions, TLR4 expression is significantly upregulated in IECs in both CD and UC, which compromise the hyporesponsiveness of IECs and amplify inappropriate immune response[69,70]. NOD2 polymorphisms in IECs are well known to be associated with an impaired intracellular microbial recognition and consequent long lasting nuclear factor kappa B (NF-kB) activation,which leads to the inflammation in CD[71,72]. Recent studies also revealed that normal IECs act as non-professional antigen presenting cells preferentially activating CD8+regulatory T cells which result in a suppressed immune response. This occurs through a combination of the non-classical MHC class I molecule, CD1d and the costimulatory molecule glycoprotein gp-180 (CD8 ligand)[73-75](Figure 1C). In contrast, the amount of CD8+ regulatory T cells was significantly decreased in IBD patients, which might be due to the defective expression of gp-180 on IECs[76]. Furthermore, IECs from IBD patients preferentially activate CD4+ T cell to proliferate and secrete IFN-γ[73].
CURRENT STATUS OF THERAPEUTIC APPROACHES IN IBD
Although IBD is a spectrum of complex multifactorial disorders and recent studies have unraveled some aspects of the critical role of intestinal epithelial dysfunction in the pathogenesis of IBD, current therapeutic strategies and targets are mainly focused on suppression of immune cell (T cell) activation, proliferation, pro-inflammatory cytokine gene expression, migration/trafficking, and resistance to apoptosis[77].Meanwhile, several drugs or therapies have been reported to promote epithelial barrier functions such as mucin production or integrity maintenance. However, only a few therapies specifically and directly target the barrier dysfunction of IECs. Thus,there is still a large unmet need for novel IBD drug discovery which potentially could be achieved based on the intestinal enteroid/organoid-culture system.
Current therapeutic approaches in IBD focused on immune cells
The Majority of current or emerging IBD therapies, at least in part, are based on suppression of pro-inflammatory cells or pathways. Classical anti-inflammatory drugs (5-Aminosalicylate, corticosteroid) inhibit pro-inflammatory cytokine production in immune cells by NF-kB inactivation[77,78]. Classical immunosuppressive agents also act through similar mechanisms. Azathioprine and 6-mercaptopurine are metabolized to suppress small GTPase RAC1, leading to inactivation and apoptosis of CD4+ T cells[79]. Cyclosporine-A and tacrolimus treatment modulate pro-inflammatory cytokine transcription and T cell survival in UC by blocking calcineurin and inducing apoptosis[80,81]. Moreover, methotrexate has a pro-apoptotic effect on proliferating CD4+ T cells with poly (ADP-ribose) polymerase (PARP)-dependent pathway[82].
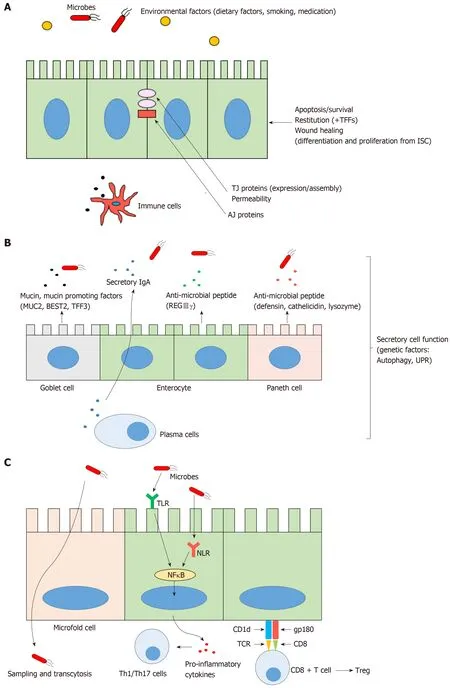
Figure 1 The normal intestinal epithelial barrier functions. A: Physical barrier function; B: Biochemical barrier function; C: Innate immune barrier function. TFFs:Trefoil factors; ISCs: Intestinal stem cells; TJ: Tight junction; AJ: Adherens junction; MUC2: Mucin2; BEST2: Bestrophin2; TFF3: Trefoil factor3; REGIIIγ: Regenerating islet-derived protein IIIγ; UPR: Unfolded protein response; TLR: Toll like receptor; NLR: Nod like receptor; NF-kB: Nuclear factor kappa B; Th: T helper; Treg:Regulatory T cells; gp180: Glycoprotein-180; CD: cluster of differentiation; TCR: T cell receptor.
Several current or emerging biologic therapies are based on blockade of proinflammatory cytokines produced by various immune cells. Anti-TNF agents(infliximab, adalimumab, golimumab, and certolizumab pegol) specifically neutralize TNF and are effective in both induction and maintenance of remission in UC and CD[83]. Furthermore, the combination therapy of azathioprine and an anti-TNF agent has dramatically reduced the need to use corticosteroids for IBD treatment[84].However, only 30-50% of patients achieve clinical and mucosal remission and significant number of initial responders experience loss of response due to antibody formation or increased drug clearance rates[85]. Recently approved ustekinumab(antibody against IL-12/IL-23 p40 subunit[86]) for CD therapy effectively suppress type 1 and type 17 T helper (Th1, Th17) cell responses which are increased in CD or UC.Approximately 30% of moderate to severe CD patients who failed to respond to anti-TNFs achieved clinical remission when they were treated with ustekinumab for 6 weeks, but half of initial responders lost response at week 44[5]. IL-6 has multiple proinflammatory effects including inhibition of apoptosis in mucosal T-cells and is a logical target for treating CD. IL-6 can bind to cells lacking the membrane-bound IL-6 receptor (IL-6R) by forming a complex with the soluble IL-6R, and subsequently induce anti-apoptotic response via IL-6/STAT3 pathway[87]. Recently, a novel anti-IL-6 antibody (PF-04236921) induced significantly higher clinical responses and remissions in refractory patients with moderate-to-severe CD following anti-TNF therapy. The overall benefit/risk profile for this drug appears to be acceptable but the observations of gastrointestinal perforation and abscess require careful consideration during future clinical development[88]. IL-6R blockade was also effective in suppressing inflammatory activity in CD. A phase 1 trial using an anti-IL-6R monoclonal antibody(tocilizumab, previously known as MRA) showed higher response and remission rates in CD patients as compared with the placebo group[89].
Moreover, there are new small molecules which inhibit intracellular proinflammatory pathways targeting cytokine signaling or transcription factors. Several Janus kinase inhibitors (JAK inhibitors: tofacitinib, filgotinib) block intracellular activation of Janus kinases which mediate cytokine signaling with respect to proliferation, survival, activation in immune cells. Patients with active UC treated with tofacitinib (JAK1/2/3 inhibitor) were more likely to have clinical response and remission than those receiving placebo in a phase II trial[90]. In vitro, tofacitinib inhibited IL-12-dependent Th1 differentiation, IL-4-dependent Th2 differentiation,and also impaired the differentiation of Th17 cells in response to IL-1β, IL-6, and IL-23[91]. Additionally, a novel ROR-γt (RAR-related orphan receptor-γt) inhibitor transiently and selectively reduced Th17 cell differentiation and cytokine production from Th17 cells but preserved group 3 innate lymphoid cells (ILC3s) which provide tissue protection in the intestine[92]. GATA3 is a transcription factor which is increased in UC patients and induces pro-inflammatory cytokines in T cells. Rectal delivery of GATA3-specific DNAzyme inhibited GATA3, and subsequently reduced Th2 cytokine (IL-6, IL-13) production in lamina propria mononuclear cells in mouse colitis[93].
Rather than targeting cytokines and immune cells which are already present in inflamed sites as above, other drugs inhibit trafficking or homing of activated T cells toward the inflamed gut. Several Integrin inhibitors (anti-α4β7 vedolizumab, anti-β7 etrolizumab) have already shown their clinical efficacies in IBD therapy[6-8,94,95].Moreover, a recent study developed a small molecule inhibitor of chemokine receptor CCR9 (CCX282-B) which is expressed in leukocyte and plays a key role for leukocyte homing to the gut mucosa[96,97]. Meanwhile, other groups are targeting adhesion molecules which are expressed in vascular endothelial cells. A novel MAdCAM-1 (gut specific ligand for integrin α4β7) antibody increased β7+ T cells and CCR9 gene expression in the peripheral blood of patients with CD in a phase 2 trial, which indicates that anti-MAdCAM-1 antibody successfully can prevent the influx of immune cells into the gut[98,99]. Alicaforsen is a human ICAM-1 antisense oligonucleotide which inhibits ICAM-1 production in vascular endothelial cells, and thereby blocks leukocyte trafficking to the mucosa[100]. Another approach regarding immune cell trafficking in IBD involves sphingosine-1-phosphate (S1P) signaling.S1P/S1P receptor signaling controls the egress of lymphocytes from lymph nodes to circulation by activating NF-kB and STAT3 transcription factors. The S1P receptor agonist/functional antagonist (ozanimod), which sequesters peripheral lymphocytes,resulted in a higher clinical remission rate in UC[101-103].
Finally, several emerging IBD therapies focus on activation of anti-inflammatory mechanisms rather than inhibition of pro-inflammatory mechanisms. Normally, antiinflammatory TGFβ/SMAD3 signaling downregulates NF-kB transcription. Under IBD conditions, SMAD7 proteins are upregulated, and thereby inhibit TGFβ/SMAD3 signaling, leading to uncontrolled inflammation. The novel anti-SMAD7 oligonucleotide (Mongersen) interferes with this inhibitory process by degradation of the SMAD7 mRNA. It subsequently resulted in significantly higher rates of clinical remission in CD patients[104]. Moreover, mesenchymal stem cells (MSCs) have been known to exert potent immunomodulatory functions on antigen specific T cells in CD patients through paracrine and cell-to-cell contact mechanisms[105]. Subsequently,various clinical trials have already shown anti-inflammatory and fistula closure effects of the MSCs in fistulizing CD patients[77,106]. In addition, T cell transplantation(regulatory T cells) in refractory CD patients, was well tolerated and induced significant reduction in the Crohn’s disease activity index[107]. Table 2 summarizes current therapeutic approaches in IBD focused on immune cells.
Current therapeutic approaches in IBD focused on IECs
A few studies have reported several drug candidates which might be helpful to correct the physical barrier dysfunction of IECs by promoting epithelial cell proliferation or TJ assembly. IL-22 is known to promote epithelial cell proliferation by acting directly on ISCs through STAT3 activation[108-110]. T cells isolated from patients with IBD produce high levels of IL-22 binding protein (IL-22BP), which inhibits IL-22.A recent study showed that the mucosal healing effect of anti-TNFα therapy may act in part by blocking IL-22BP[111]. Also, oral tauroursodeoxycholate (TUDCA) restores ER function by improving UPR and ameliorates colitis in mice[112]. Epidermal growth factor (EGF) is indispensable for proliferation of ISCs and is used as an important growth factor in intestinal enteroid/organoid cultures[21,113]. A preliminary study suggested that EGF enemas are an effective treatment for active left-sided UC[114]. The modulation of TJs can also be a promising target by increasing epithelial integrity. An octapeptide (AT-1001), which inhibits the disassembly of TJs by interfering with cytoskeletal rearrangement, appeared to reduce epithelial barrier dysfunction in celiac disease[115]. Celiac disease shares some common immune pathogenesis with IBD,including a dysregulated intestinal epithelial permeability[116], which suggests that AT-1001 might be considered as a novel IBD drug. Moreover, several existing drugs(vitamin D, estradiol, and metformin) also have shown to reduce epithelial permeability and to alleviate mucosal inflammation in IBD mouse models[117-119].
There have been other approaches to correct the biochemical dysfunctions or innate immune dysfunctions of IECs. However, most of them focus on mucus dysfunction of IECs. Several therapeutic approaches such as prostaglandin E2 (PGE2) receptor EP4 agonist, butyrate (short chain fatty acid), and cathelicidin have shown to induce mucus production in IECs as well as to reduce colitis in mice[120-124]. A novel mucin component therapy (phosphatidylcholine) has shown to produce a significant improvement in disease activity in UC patients[125,126]. However, phase III clinical data are still missing. In addition, two existing drugs (TLR7 ligand imiquimod, probiotic Escherichia coli Nissle) stimulate the production of antimicrobial peptides in IECs, and thus result in reduced colitis activity in mice[127,128]. Finally, Etrolizumab which was mentioned above as an anti-β7 integrin inhibitor, has a second effect with respect to barrier dysfunction. This drug blocks the interaction between αE (CD103) on intestinal T cells with E-cadherin on IECs, and subsequently inhibits the intestinal retention of CD4+ and CD8+ T cells in inflamed sites of IBD. This mechanism is considered to be one of the reasons why etrolizumab was more potent than vedolizumab in reducing accumulation of T cells in the inflamed gut[129]. This result also underscores the need of developing IBD drugs specifically targeting innate immune dysfunction of IECs(summarized in Table 3).
CURRENT ENDEAVORS TO IDENTIFY A NOVEL THERAPEUTIC TARGET FOR IBD USING INTESTINAL ENTEROIDS/ORGANOIDS
Since the group of Hans Clevers and Toshiro Sato successfully generated intestinal enteroids/organoids from intestinal crypts, many studies have been performed using human or mouse intestinal enteroids/organoids not only to investigate general intestinal stem cell physiology but also to identify the pathophysiology of specific intestinal diseases, such as colorectal cancer[130,131], infectious diarrheal diseases[38],metabolic diseases[132,133], and genetic disorders[134]. Studies to date to identify a novel therapeutic target for IBD have only used human ISC not iPSC derived intestinal enteroids/organoids. Although several studies have explored the pathogenesis of IBD using intestinal enteroids/organoids with a goal of identifying novel drug targets, the number of successful studies is small relating to the specific IBD drug development.In this section, we provide a summary of the present status of IBD drug research using intestinal enteroids/organoids.
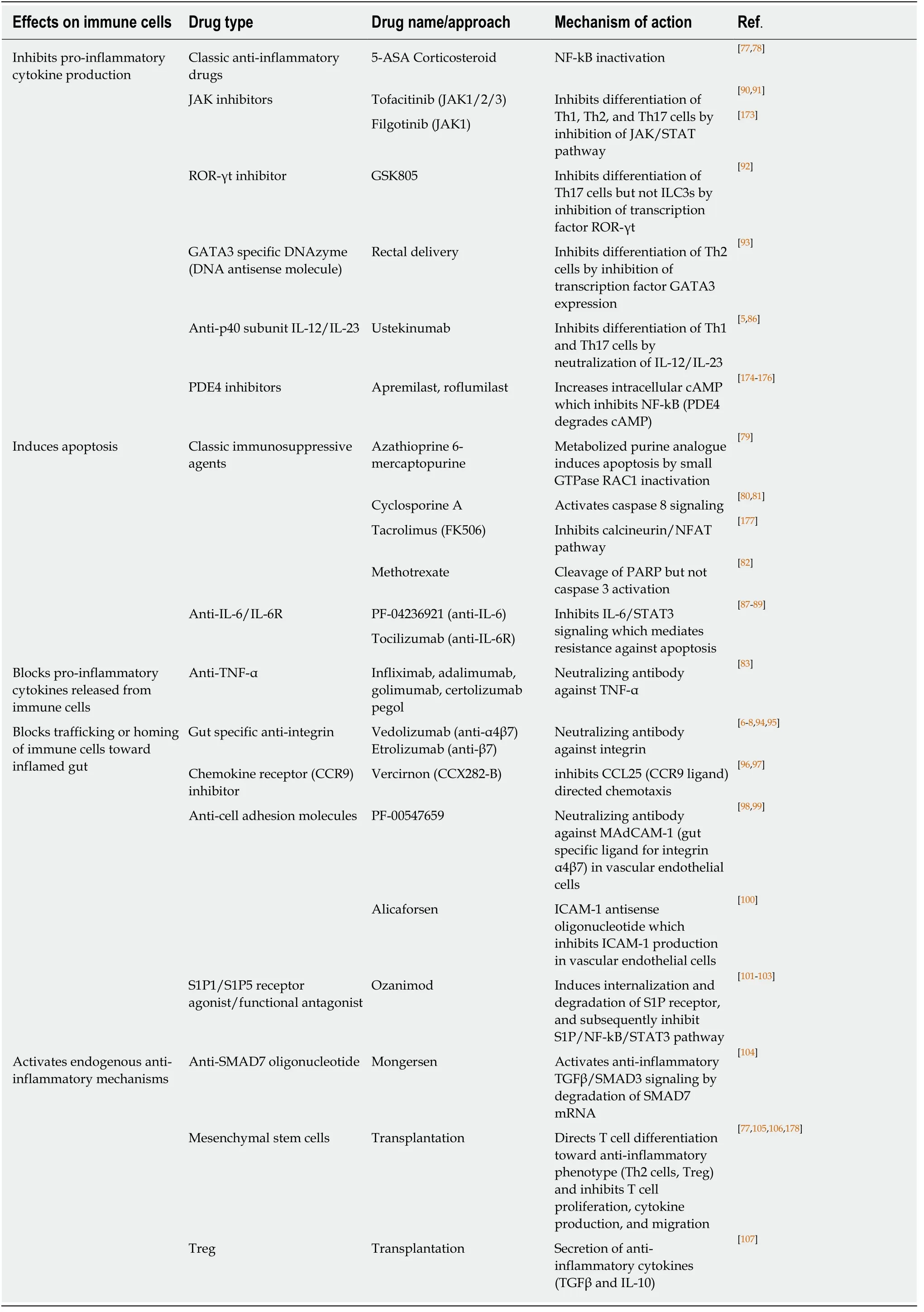
Table 2 Current therapeutic approaches in inflammatory bowel diseases focused on immune cells
Comparative study among intestinal enteroids/organoids derived from control and IBD patients for discovery of novel target molecule
A previous study found a novel drug target IL-1 decoy receptor (IL-1R2) which is highly expressed in IECs in UC patients in remission compared with those in patients with active UC and in healthy controls. This decoy receptor blocks the IL-1β dependent inflammatory mechanism. The addition of an anti-IL-1R2-blocking antibody significantly enhanced IL-1β-dependent transcription of pro-inflammatory chemokines such as CXCL1, CXCL2, and CCL20 in intestinal enteroids/organoids from UC patients in remission. However, there was no significant difference in intestinal enteroids/organoids from healthy controls. Thus, IL-1 R2 may be a novel drug target to promote remission in UC[135].
Rare subtypes of IBD with very early-onset in early childhood (VEO-IBD) are more likely to be associated with specific genetic mutations, especially associated with immune dysfunction, including in IL-10, IL-10R, XIAP, NCF2, or TTC7. These do not occur in IBD in older children[136]. Using enteroids derived from these young patients can lead to the discovery of compounds which are able to normalize the consequence of those mutations. For example, intestinal enteroids grown from children with multiple intestinal atresia, a rare congenital disease with IBD-like features by TTC7A mutations, displayed an inversion of apical-basal polarity of the epithelial cells that was reversed by Rho kinase inhibitor[137]. More recently, whereas TRAIL stimulation induced caspase-3 cleavage and cell death in intestinal enteroids derived from healthy donors, caspase-8 deficient intestinal enteroids derived from a VEO-IBD patient were unresponsive to TRAIL, revealing caspase-8 deficiency as a novel cause for VEO-IBD associated with defective epithelial cell death response[138].
Recently, several studies have compared the profiles of gene expression and DNA methylation in intestinal enteroids/organoids derived from control and IBD patients[14,27,28]. A group of genes were differentially regulated in the intestinal enteroids/organoids of UC patients, including genes associated with antimicrobial defense (LYZ, PLA2G2A) and with mucus secretory functions (ZG16, CLCA1)compared with intestinal enteroids/organoids of non-IBD subjects[14]. These genes which are associated with epithelial barrier dysfunction might be potential drug targets for future IBD therapy.
Development of intestinal enteroid/organoid-based IBD model by administration of conditions mimicking IBD
In the IBD environment, several luminal and lamina propria factors such as microbes,cytokines, drugs, and metabolites contribute to the epithelial dysfunction, and subsequently result in the initiation and progression of IBD. Therefore, adding these conditions to intestinal enteroid/organoid culture models might more closely recapitulate the IBD pathophysiology. Previous studies have added diverse IBD related factors (cytokines, bacterial components, metabolites, and niche factors for ISCs) to organoid cultures to model the epithelial barrier dysfunctions in IBD[19,109,139-141].
In terms of physical barrier dysfunction, the target molecules which contribute to epithelial apoptosis or regeneration after injury have been studied. For example, IFNγ strongly and rapidly induces degranulation and extrusion of Paneth cells in intestinal enteroids/organoids. Furthermore, prolonged stimulation with recombinant IFN-γ (1 ng/mL) reduced intestinal enteroid/organoid growth and caused progressive loss of Paneth cells due to apoptosis[139]. Therefore, this study not only showed that the intestinal enteroid/organoid culture is a useful model to investigate Paneth cell dysfunction in IBD, but also suggested that the pharmacologic modulation of IFN-γ signaling may allow a new strategy to correct Paneth cell dysfunction and survival in IBD.
TNF-α is another pro-inflammatory mediator that plays an integral role in IBD pathogenesis. This cytokine was used to disrupt intestinal enteroids/organoids to recapitulate epithelial apoptosis in IBD[19]. Meanwhile, other studies have found several niche factors for ISCs which may act to promote epithelial proliferation and regeneration after injury. The epithelial regeneration is critical to maintain the physical barrier function after intestinal injury. Previous studies have already reported that several niche factors for ISCs such as Wnt, Notch, and EGF support Lgr5+ ISCs for normal epithelial maintenance[21,142]. A recent study found that IL-22,which is produced from ILCs after tissue injury, also augmented the growth of intestinal enteroids/organoids and proliferation of ISCs via the IL-22/STAT3 phosphorylation pathway. Treatment with IL-22 promoted the recovery of ISCs and epithelial regeneration in injured enteroids after irradiation. Thus, IL-22/STAT3 signaling might be a potential drug target to promote mucosal healing in IBD[109].Similarly, a co-receptor for IL-6 (gp130) promoted the regeneration of intestinal enteroids/organoids by activating YAP and Notch signaling, which underscores that normalization of IL-6 signaling, rather than complete inhibition, may be a future therapeutic approach to improve epithelial regeneration in IBD[143]. In addition,bacterial metabolites, including short-chain fatty acids (SCFAs), increased the size and the number of buds per enteroids/organoids, which supports that commensal bacteria reinforce epithelial barrier function by inducing proliferation of IECs with SCFAs production[11,140]. Following disruption of the epithelial barrier, a transient,specialized repair cell type, wound-associated epithelial (WAE) cells rapidly migrate and seal the epithelial defects (termed restitution)[144]. A previous study using intestinal enteroids/organoids demonstrated that PGE2/EP4 signaling also promotes epithelial regeneration after injury by driving ISCs to differentiate toward WAE cells[145].

Table 3 Current therapeutic approaches in inflammatory bowel diseases focused on intestinal epithelial cells
Furthermore, there are several studies that model the biochemical- and innate immune-epithelial barrier functions which are relevant to IBD. Colonic enteroids/organoids (termed colonoids) derived monolayers were differentiated to study the interaction of Enterohemorrhagic Escherichia coli (EHEC) and human colonic epithelium,which mimic some aspects of host-pathogen interaction in IBD. Colonized EHEC reduced mucus production of colonoids and destroyed the mucus layer[146]. EHEC strain O157:H7 is known to block the IFN-γ/STAT1 pathway in epithelial cells[147].This mechanism is very similar to the pathogenic feature of adherent invasive Escherichia coli (AIECs) isolated from CD patients, which has been implicated in the pathogenesis of CD and is known to suppress the IFN-γ/STAT1 pathway in IECs.This suggests this bacteria might induce an inappropriate anti-microbial response in CD[148,149]. Therefore, these human enteroid monolayer cultures might also be used as a relevant pathophysiologic model that allow the study of the role of AIECs in the context of biochemical barrier dysfunction in IBD. CD is thought to result from an inappropriate inflammatory response directed toward commensal enteric microflora and specific bacterial antigens targeted by this immune response have been studied.Especially, bacterial flagellin (TLR5 ligand) appears to be one such target of the CDassociated immune response[150-152]. Flagellin-specific serum IgG was elevated in patients with CD, but not in patients with UC or in controls, which suggests that flagellin is an antigenic target of the elevated adaptive immune response in CD[150-152].Flagellin is known to be associated with pro-inflammatory IEC activation as well as pro-fibrotic myofibroblast activation[153-155]. In a recent study, human colonoids from healthy individuals were exposed basolaterally to diverse TLR ligands including(TLR2, TLR4, and TLR5) to investigate the pro-inflammatory response. Flagellin stimulated an increase in both IL-8 and TNF-α transcripts in colonoids in contrast to TLR2 and TLR4 ligands which failed to induce those cytokines[155]. In this study, proinflammatory response to flagellin in colonoids from CD patients was not compared to those from healthy individuals. It remains to be determined what the roles of the flagellin and TLR5 receptor are in colonic epithelial cells in CD.
PGE2 is a well-known mediator that promotes diarrhea in IBD by directly activating chloride (Cl-) and fluid secretion through the CFTR on the apical surface of IECs[156-158]. The expression of PGE2 is upregulated in the inflammatory mucosa of IBD patients, where other inflammatory cytokines are also abundant. In a recent study using intestinal enteroids/organoids established from uninflamed mucosa of CD patients, short-term treatment with cytokines such as TNF-α, IL-1β, IL-6, or IFN-γ alone failed to induce a secretory response of enteroids, while PGE2 alone induced enteroid swelling. Furthermore, none of the tested cytokines was able to affect the PGE2-induced enteroid swelling, which suggests the possibility that PGE2 may act as a robust and direct mediator of fluid secretion from IECs and the PGE2 level might determine the local secretory response of IECs without the involvement of other inflammatory cytokines[159]. However, further comparative studies using enteroids from the inflamed mucosa of IBD patients are required to elucidate what the specific role of PGE2 is in the pathophysiology of inflammatory IBD diarrhea.
More recently, to describe long-term epithelial responses to inflammatory stimulation mimicking chronic IBD, a mixture of cytokines and bacterial components were added to the intestinal enteroid/organoid model for 60 weeks. In this study,long-term inflammation led to sustained NF-kB activation and reactive oxygen species (ROS) production even 11 weeks after the removal of the inflammatory mixture. A perpetual NF-kB activation in IECs has been implicated in innate immune dysfunction of IECs in IBD[71,72]. Therefore, this long-term intestinal enteroid/organoid model with sustained NF-kB activation has the potential to recapitulate the perpetual inflammatory activity in mucosa of IBD. Additionally, this result suggests that the long-term inflammatory stimulation might transform intestinal enteroids/organoids into a stressed cellular state, which might be related to the etiology of colitisassociated cancer in UC[48]. In the context of long-term complication of IBD, our previous study used synergistic treatment of TNF-α and TGFβ to generate an intestinal enteroid/organoid model recapitulating epithelial mesenchymal transition(EMT), which might provide a cellular source of myofibroblasts and contribute to intestinal fibrosis in CD[160].
Development of intestinal enteroids/organoids-based IBD model by co-culturing with other cell components
In IBD pathogenesis, IECs interact with diverse types of cells including immune cells,mesenchymal cells and neuronal cells. Thus, co-culturing intestinal enteroids/organoids and other cell components can provide a more complex model which includes cell-to-cell interactions, to more closely elucidate the in vivo pathophysiology of IBD. So far, the number of studies co-culturing enteroids/organoids with other cells to specifically investigate IBD pathogenesis is limited. Most co-culture studies have examined the role of other cells for the growth of intestinal enteroids/organoids which is associated with physical barrier function of IECs. For example, to evaluate the role of ILC3s for IECs, lamina propria ILC3s capable of producing IL-22 were cocultured with intestinal enteroids/organoids. ILC3s markedly increased the size of intestinal enteroids/organoids, and this phenomenon was blocked by an anti-IL-22 neutralizing antibody. Therefore, this co-culture model revealed that immune cells are able to activate ISCs to promote proliferation[109]. Moreover, as a potent supporter of intestinal barrier, the diverse roles of IL-22 producing ILC3s have been reported including promoting epithelial proliferation and inducing anti-microbial peptide and mucin production. In the majority of human studies, the differential frequencies of ILC3 subsets are critically involved in the pathogenesis of CD. Protective NKp44+, IL-22 producing ILC3 subset was found to be decreased whereas the frequency of the IL-17- and IFN-γ-producing CD56-subset of ILC3 was increased in the intestine of CD patients[110,161]. Therefore, this co-culture model of intestinal enteroids/organoids and ILC3 might provide a valuable tool to study IECs-ILC3 interaction in IBD. Another epithelial cell type, tuft cell, has been shown to express the doublecortin-like kinase 1 protein (DCLK1) as a specific marker. DCLK1+tuft cell cannot survive under standard organoid culture condition. A recent study established a co-culture model of primary neurons and intestinal enteroids/organoids and found that nerves can prolong the survival of DLCK+tuft cells in vitro. Surprisingly, they also demonstrated that DLCK+tuft cells give rise to poorly differentiated colorectal cancer after inflammatory injury,suggesting that DLCK+tuft cells might serve to initiate CAC in IBD[162]. Thus, this system should be further developed as a CAC model in IBD.
Noel et al[163]generated a human macrophage-intestinal enteroid/organoid coculture system and showed that macrophages (M0) enhance physical barrier function and maturity of intestinal organoid monolayers as demonstrated by trans-epithelial electrical resistance and cell height. Although the two effector macrophages (M1 and M2) which are involved in IBD pathogenesis, were not included in this study, this system can be further developed to investigate the interaction between IECs and macrophages in the context of innate immune function of IECs[163]. Emerging evidences have suggested that mesenteric adipose tissue may play a critical role in the inflammatory and fibrotic pathogenesis in CD. In CD, longitudinal ulcerations are located primarily along the mesenteric attachment and the “creeping fat” is known to be implicated in muscular hypertrophy and subsequent stricture formation in CD[164,165]. Recently, the co-culture of intestinal enteroids/organoids and adipocytes revealed a reciprocal inflammatory activation between these cell types, which suggests that adipocytes can be a promising drug target for CD treatment[166].
THE OBSTACLES TO OVERCOME IN USING INTESTINAL ENTEROIDS/ORGANOIDS AS AN IBD RESEARCH TOOL
Although intestinal enteroid/organoid culture system has many advantages over the conventional IEC models, there are multiple issues to resolve before researchers use intestinal enteroids/organoids as a platform to develop novel IBD drugs.
The drawbacks of intestinal enteroid/organoid culture conditions
Cost is a big drawback of intestinal enteroid/organoid cultures. Currently, a significant amount of expensive Matrigel, which is derived from mouse sarcoma cells,is required to propagate intestinal enteroids/organoids. The culture medium contains a myriad of growth factors which have multiple sources such as conditioned media from cell lines and purified recombinant proteins. Both Matrigel and growth factors from conditioned media contain varying amounts of xenogeneic and undefined components, along with the potential risk of pathogen/immunogen transmission and batch-to-batch variability[38,167,168].
Considering the recent progress in establishing human enteroids as a model of infectious diarrheal diseases, human enteroid/organoid culture conditions might provide a relevant pathophysiologic model to study microbiota-epithelium interactions[38,146]. However, to develop a more reliable model, ways to co-culture the primarily anaerobic gut microbiota, which consists of many bacterial species, with human enteroids in anerobic culture conditions without damaging the enteroids is a significant challenge to be overcome[9]. Moreover, intestinal enteroid/organoid culture systems lack physiological luminal and blood flow and the repetitive contractions of peristalsis, which are known to result in altered gene expression and cell function/morphology[168]. In this regard, an emerging microfluidic chamber system combining peristalsis-like stretch with apical and basolateral liquid flow (gut-on-achip) might be a way to overcome these drawbacks of enteroid/organoid cultures.Most recently, a novel microfluidic device with a transluminal hypoxia gradient was developed for co-culturing a complex living human gut microbiome, including obligate anaerobes with human intestinal epithelium, that until now has only included Caco-2 cells[169].
The issues related to intestinal enteroids/organoids derived from patients with IBD
In the mucosa of active IBD, excessive cell death is observed in the ileal and colonic epithelium due to a highly inflammatory and pro-apoptotic milieu within the lamina propria[170]. Therefore, the number and quality of isolated intestinal crypts and ISCs might be too low to regularly form intestinal enteroids/organoids as well as to maintain long-term culture. In particular, the intestinal crypts isolated from actively inflamed lesion in CD, in our experience, showed low yields of intestinal enteroid/organoid formation. Moreover, a recent study mentioned the difficulty of generating intestinal enteroids/organoids, even from the normal mucosa of UC patients due to crypt distortion[47]. These observations suggest that the current protocols to isolate and culture intestinal enteroids/organoids might not be sufficient for generating enteroid/organoid banks of actively inflamed IBD. A few studies reported that the intestinal enteroid/organoid-forming capacity is not lower in the crypts or ISCs derived from patients with active CD than in those derived from patients with inactive IBD or from healthy controls[14,27,171]. However, the severity of inflamed mucosa which was used to isolate intestinal enteroids/organoids was not standardized among those studies, which might affect the intestinal enteroid/organoid-forming capacity. The number of published studies which have used intestinal enteroids/organoids derived from actively inflamed lesions of IBD,especially CD, is still very limited.
As a surrogate of IECs in IBD, whether the intestinal enteroids/organoids derived from IBD patients will resemble the inflammatory phenotype of the tissues of origin on the long-term culture is another issue to be elucidated. Although several studies have suggested that the intestinal enteroids/organoids from patients with IBD retain the disease-specific DNA methylation and gene expression changes which might be imprinted in ISCs, a recent study showed that the expression of IFN-γ and IL-1β appeared to be significantly lower in intestinal enteroids/organoids derived from patients with IBD compared with biopsies used to form these intestinal enteroids/organoids. This suggests that inflammatory phenotype at the mRNA level in biopsies may not be propagated to intestinal enteroids/organoids, and a proinflammatory stimulation might be required for sustained expression of the inflammatory phenotype. However, in this study, whole tissue with inflammatory cells was compared to enteroids without inflammatory cells. Thus, status of inflammation in IBD enteroids over passages needs determination. Furthermore,considering that there was a trend that the expression of IFN-γ in UC inflamed organoids was higher than in control organoids and that limited numbers of IBD patients (n = 3-7 for each group) were enrolled to isolate enteroids/organoids in this study, further studies are needed to elucidate whether the transcription profiles of enterods/organoids will remain identical or similar to those of the inflamed tissues of origin in IBD[48,171].
CONCLUSION
Although IBD is a multifactorial and heterogeneous disease, the therapeutic approaches over the last decades have been intensively focusing on immune cells and anti-inflammatory mechanisms. Moreover, despite the therapeutic advances including novel biologics, a significant number of IBD patients still do not achieve long-term remission and mucosal healing, which are very important for the quality of life and the prognosis of these patients.
The emerging evidence has underscored that the intestinal epithelium has a crucial role in maintaining mucosal homeostasis and regulating immune cells as a physical,biochemical and innate immune barrier. Moreover, epithelial barrier dysfunction is regarded as a critical mechanism in development and perpetuation of IBD. However,as described through this review, the current therapeutic approaches in IBD focused on epithelial barrier dysfunction are still limited. Compared with conventional IEC cultures, a novel intestinal enteroid/organoid culture might provide advantages to recapitulate the in vivo physiology of human intestinal epithelium, which makes it a promising tool for modeling the pathophysiology of IBD. However, the number of published studies using the intestinal enteroid/organoid model to specifically identify drug targets for IBD treatment is thus far small. More diverse therapeutic approaches may ultimately overcome the limitations of current therapeutic options.Therefore, IBD drug research focusing on epithelial dysfunction with a novel intestinal enteroid/organoid system should be evaluated as a future way to develop additional therapeutic approaches of IBD.
杂志排行

World Journal of Gastroenterology的其它文章
- Exhaled breath analysis in hepatology: State-of-the-art and perspectives
- Miniature gastrointestinal endoscopy: Now and the future
- Issues and controversies in esophageal inlet patch
- Role of hepatocyte nuclear factor 4-alpha in gastrointestinal and liver diseases
- G protein-coupled estrogen receptor in colon function, immune regulation and carcinogenesis
- Helicobacter pylori and cytokine gene variants as predictors of premalignant gastric lesions