微囊藻毒素LR免疫原及包被抗原的合成与鉴定
2019-08-20何丹刘媛李先保龚航王丽郝佳张晓帅徐重新张存政刘贤金
何丹 刘媛 李先保 龚航 王丽 郝佳 张晓帅 徐重新 张存政 刘贤金


摘要:微囊藻毒素LR(MC-LR)分子量小于1 000 u,为半抗原,需要与载体蛋白偶联后才能刺激免疫应答用于抗体制备。本研究利用碳二亚胺法将MC-LR分别与钥孔血蓝蛋白(KLH)和牛血清白蛋白(BSA)进行偶联,获得免疫抗原(MC-KLH)和包被抗原(MC-BSA)。采用紫外扫描光谱、SDS聚丙烯酰胺凝胶电泳(SDS-PAGE)、基质辅助激光解吸电离飞行时间质谱(MALDI-TOF MS)等3种方法对2种人工抗原进行鉴定,并对小鼠进行免疫。用间接非竞争酶联免疫吸附测定法(ELISA)和竞争ELISA测定了抗血清效价和敏感度。结果表明,紫外扫描光谱法可观察到2种人工抗原的紫外吸收谱与载体蛋白相比发生改变。通过SDS-PAGE法可以观察到MC-BSA分子量比BSA增大。另外质谱法测定的MC-BSA的相对分子量为76 515.84,计算得到的MC-LR与BSA的偶联比为10 ∶1。MC-KLH免疫小鼠后,抗血清效价最高达到1 ∶32 000倍,MC-LR对鼠多抗血清的抑制中浓度(I50)为0.53 μg/mL。成功合成了MC-LR的免疫原和包被原为其单克隆抗体的制备奠定了基础。还比较了3种鉴定方法的适用性和优缺点,为其他人工抗原的鉴定方法提供了有益的参考。
关键词:微囊藻毒素LR;抗体;免疫原;包被原;鉴定
中图分类号:X52 文献标志码: A
文章编号:1002-1302(2019)09-0226-05
微囊藻毒素(microcystins,简称MCs)是一种在蓝藻水华污染中出现频率最高、产生量最大和造成危害最严重的藻毒素种类[1],其主要结构为环D-丙氨酸-R1-R2-赤-β-甲基-D-异天冬氨酸-L-Z-adda-D-异谷氨酸-N-甲基脱氢丙氨酸,其中有2个可变的L-型氨基酸(R1和R2),这2种L-型氨基酸的不同组合更替及其他氨基酸的去甲化衍生出众多的毒素类型,至今已发现80种以上的MCs异构体[2-4](图1)。其中微囊藻毒素LR(MC-LR)是已知毒性最强、急性危害最大的一种淡水蓝藻毒素,具有多器官毒性、遗传毒性和致癌性[5]。世界卫生组织及许多发达国家制定的引用水标准和规范中规定,MC-LR的含量不得超过 1.0 μg/L,我国生活饮用水卫生标准也规定饮用水中的 MC-LR不得超过1.0 μg/L[6]。
目前MC-LR的检测方法主要包括液相色谱法、免疫分析法、磷酸酶抑制法、细胞毒性法和生物分析法等[7-15]。免疫分析法因其灵敏度高、检测通量大等优点成为MC-LR最常用的筛选检测方法,其核心试剂抗体的制备国内外已有一些文献报道[16]。然而目前商品化的MC-LR抗体仍然非常有限,且质量参差不齐。进口的MC-LR单克隆抗体主要为英国Abcam公司的产品,售价约达3 000元/mg。国内仅有中国水生生物研究所、江苏省微生物研究所和清华大学等少数几家单位研制的MC-LR单、多克隆抗体。现有的商品化MC-LR抗体难以完全满足巨大市场检测需求和科研使用。因此,加快MC-LR单克隆抗体的研制和自主知识产权的快速检测技术产品的研发,为解决我国特别是江苏地区太湖蓝藻暴发水体污染监测等社会、环境问题意义重大。
由于MC-LR分子量为995.17 u,属于半抗原,必须要和载体蛋白偶联才能具有免疫原性,从而获得抗体。因此,合成性能良好的免疫原及包被原是制备抗体和建立检测方法的关键。本研究利用碳二亚胺法将MC-LR与载体蛋白钥孔血蓝蛋白(KLH)、牛血清白蛋白(BSA)分别进行偶联,作为免疫原和包被原,并用紫外扫描光谱、十二烷基硫酸钠聚丙烯凝膠电泳(SDS-PAGE)和基质辅助激光解吸电离飞行时间质谱(MALDI-TOF MS)等3种方法进行鉴定。
1 材料与方法
1.1 主要材料
微囊藻毒素LR,购自瑞典A1exis Biochem公司。6~8周龄雌性Balb/c小鼠,购自江苏省扬州大学比较医学中心。钥孔血蓝蛋白、牛血清白蛋白、50%聚乙二醇(PEG)溶液(分子量为1 450 ku)、弗氏完全和不完全佐剂,均购自美国Sigma公司。N,N-二甲基甲酰胺(DMF),购自无锡中西药品器械集团有限公司。辣根过氧化物酶(HRP)标记羊抗鼠IgG,购自南京康维生物科技有限公司。1-乙基-3(3-二甲氨丙基)碳二亚胺盐酸盐(EDC·HCl)、N-轻基琥珀酰亚胺(NHS)、四甲基联苯胺(TMB),均购自上海丽珠东风生物技术有限公司。透析带(截留分子量为14 000 ku),购自上海华美生物工程有限公司。4×蛋白上样缓冲液、脱脂奶粉,均购自索莱宝生物科技有限公司。10%预制胶,购自南京金斯瑞生物科技有限公司。
1.2 主要仪器
酶标仪(美国Thermo),洗板机(美国Thermo),台式冷冻离心机(美国Beckman),紫外-可见分光光度计(美国PerkinElmer),微量可调移液器 (芬兰Finnpipette),超纯水净化仪(美国Millipore),分析天平(美国OHUS),磁力搅拌器(杭州仪表电机有限公司),96孔酶标板(美国CORNING),垂直板电泳仪(美国Bio-rad),MALDI-TOF/TOF 5800质谱分析仪(美国AB Sciex公司),5% CO2培养箱(美国Thermo)等。
1.3 免疫抗原和包被抗原的合成
免疫抗原(MC-KLH)的制备:MC-LR与KLH的偶联,参考Yu等的方法[17]。具体为取0.1 mg MC-LR溶解于 0.1 mL DMF,再与0.3 mL溶解有6 mg 1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸酸(EDC·HCl)和6 mg NHS的DMF溶液混合,轻微振荡并混匀,室温反应30 min后,4 ℃反应过夜,得MC-LR活泼酯衍生物(Ⅰ)。称取 3 mg KLH溶解于3 mL 0.1 mol/L pH值为9.6的碳酸盐缓冲液(CBS)中,将衍生物(Ⅰ)缓慢滴加入KLH溶液中,轻微振荡并混匀,避光室温反应2 h。将反应物移入透析袋,透析袋放在pH值为7.4的0.01 mol/L磷酸盐缓冲液(PBS)中,4 ℃下透析2 d,每天更换透析液3次,透析产物分装后于-20 ℃保存。
包被抗原(MC-BSA)的制备:将0.1 mg MC-LR溶解于25%乙醇溶液中,再加入335 μg BSA,振荡混匀。随后将溶有1 mg EDC·HCl 的200 μL水溶液逐滴加入MC-LR和BSA的混合液中,4 ℃反应过夜。制备好的包被抗原装入透析袋,在PBS缓冲液中4 ℃下透析2 d,每天更换透析液3次,分装后于-20 ℃保存。
1.4 鉴定方法
1.4.1 紫外扫描光谱法 将免疫抗原、包被抗原溶解于PBS中,使其浓度为200 μg/mL,载体蛋白KLH、BSA浓度为 1 mg/mL,半抗原MC-LR浓度为10 μg/mL,用紫外分光光度计对各物质进行紫外全波长扫描,绘制紫外吸收曲线。
1.4.2 SDS-PAGE法 配制200 μg/mL的MC-BSA和BSA,与蛋白上样缓冲液混合后,煮沸3 min。样品上样量为 5 μL/孔,蛋白marker上样量为2 μL/孔。使用10%的预制胶分离,140 V恒压下电泳70 min,通过银染法显色。
1.4.3 基质辅助激光解吸电离飞行时间质谱法 将1 μL 0.5 mg/mL PBS溶解的BSA或MC-BSA偶联物点至样品靶上,自然干燥后,再取0.6 μL CHCA基质溶液点至对应靶位上并自然干燥,用相同方法在样品靶位相邻位置点标准品。在正离子模式下选择反射方法对样品测试范围进行校准测试。反射校准物质范围为39 212±100至66 430±100;在正离子模式下选择反射方法测试样品分子量。质谱数据及图谱处理由MALDI-TOF MS产生,原始数据及图谱由4000 Series Explorer V3.5软件导出。按照以下公式计算半抗原与载体蛋白偶联比:偶联比=(偶联物分子量-载体蛋白分子量)/半抗原分子量。
1.5 动物免疫
取适量的MC-KLH与等体积完全弗氏佐剂混合,完全乳化,免疫3只6~8周龄雌性Balb/c小鼠,25 μg/只腹腔注射。3周后,取适量的MC-KLH与等体积不完全弗氏佐剂混合,乳化后皮腹腔注射,根据此方法每14 d免疫1次,共加强免疫4次。选取效价最高小鼠,用PBS溶解的25 μg MC-KLH进行腹腔注射,进行冲击免疫,3 d后取脾脏融合。
1.6 抗血清效价及灵敏度的测定
1.6.1 间接非竞争酶联免疫吸附测定法(ELISA)测定效价 (1)包被:用 2.5 μg/mL CBS稀释的MC-BSA,100 μL/孔加入酶标板,4 ℃ 包被过夜。(2)封闭:次日用含有0.05% Tween 20的PBS溶液(PBST)洗板3次,每孔加入200 μL PBS配制的体积分数为2%的脱脂奶粉,37 ℃封闭1 h。(3)加样:PBST洗板3次后,每孔加入100 μL PBS梯度稀释的鼠阳性血清,以相应稀释倍数的鼠阴性血清作为阴性对照,37 ℃温育1 h。(4)加酶标二抗:PBST洗板3次,每孔加入100 μL HRP标记的羊抗鼠IgG工作液(1 ∶5 000倍PBS稀释),37 ℃温育1 h。(5)显色:PBS洗板3次后,每孔加入100 μL现配的底物溶液(100 μL二甲亚砜溶解的TMB和25 μL 0.65% H2O2加入9 .875 mL 0.1 mol/L pH值为5.5的柠檬酸缓冲液),37 ℃显色15 min后,每孔加入50 μL 2 mol/L H2SO4终止液终止反应,在酶标仪上读取450 nm处的吸光度,以大于相应的阴性血清吸光度2.1倍的阳性血清最大稀释倍数作为抗血清效价。用方阵滴定法确定包被抗原及鼠血清的工作浓度。
1.6.2 间接竞争ELISA测定抗血清敏感度 酶标板的包被及封闭、加酶标二抗和显色步骤同“1.6.1”节。加样步骤操作如下:用PBS将MC-LR标准品稀释成100、10、1、0.1、0.01、0.001 μg/mL等6个浓度,酶标板每孔加入50 μL 不同浓度MC-LR标准品和50 μL 2倍工作浓度的鼠抗血清混合液,以50 μL PBS和50 μL 2倍工作浓度的鼠抗血清混合液作为零浓度的MC-LR。计算不同浓度MC-LR标准品对应吸光度(B)与零浓度MC-LR对应吸光度(B0)的比值,绘制MC-LR浓度与B/B0的标准抑制曲线,并计算抑制中浓度(I50)。
2 结果与分析
2.1 紫外扫描光谱法
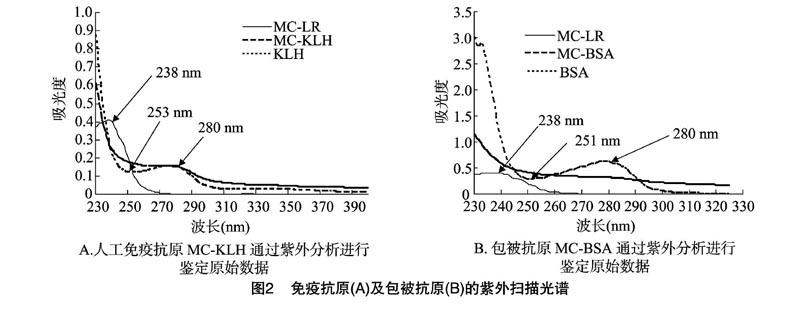
紫外扫描光谱法是免疫抗原和包被抗原鉴定的一种常用方法。它是通过比较载体蛋白、人工抗原、半抗原的紫外吸收光谱的迁移或叠加情况来分析偶联结果。图2-A显示了免疫抗原MC-KLH、半抗原MC-LR和载体蛋白KLH的紫外扫描光谱。在紫外扫描范围内,半抗原MC-LR在238 nm处具有特征吸收峰,KLH在280 nm处具有蛋白质特征吸收峰,在 253 nm处出现波谷。偶联物MC-KLH在238~280 nm范围内,紫外吸收曲线呈平滑下降趋势,没有KLH的280 nm处的特征吸收峰和253 nm处的波谷出现,这可能是由于MC-LR与KLH偶联后,2种物质的紫外吸收发生叠加导致,由此推测MC-LR和KLH偶联成功。
同样图2-B显示了包被抗原MC-BSA、MC-LR和BSA的紫外扫描光谱。BSA在280 nm處具有蛋白质特征吸收峰,在251 nm处出现波谷。偶联物MC-BSA在238~280 nm 范围内,紫外吸收曲线呈平滑下降趋势,没有BSA的280 nm处的波峰和251 nm处的波谷出现,这可能是由于 MC-LR 与BSA偶联后,两者紫外吸收发生叠加导致,由此推测MC-LR和BSA偶联成功。
2.2 SDS-PAGE法
SDS-PAGE法是鉴定蛋白分子量的经典方法。由于SDS-PAGE法的电泳迁移率主要取决于蛋白分子量的大小,可以通过比较载体蛋白和偶联物的电泳迁移率来分析是否偶联成功。如图3所示,BSA的条带在72 ku下方,符合66 ku 的BSA分子量预期,而MC-BSA的条带接近于72 ku。MC-BSA 的分子量大于BSA,因此可以推测MC-LR与BSA偶联成功。
由于试验所用的KLH蛋白来源于海洋软体动物钥孔帽贝(Megathura crenulata),该蛋白由KLH1和KLH2(350~400 ku)2个亚基聚合而成,分子量范围在400~800 ku之间。并非单一分子量纯品,而且KLH分子量过大不能适用于常规的蛋白marker及凝胶,因此未用此种方法对MC-KLH进行鉴定。
2.3 基质辅助激光解吸电离飞行时间质谱法
MALDI-TOF MS的原理是用一定强度的激光照射样品与基质形成共结晶薄膜,基质从激光中吸收能量,基质-样品直接发生电荷转移使得样品分子电离后在电场作用下加速飞过飞行管道,根据到达检测其的飞行时间不同而被检测。即依据样品质荷比(m/z)不同进行检测得到样品的相对分子质量。
试验用MALDI-TOF MS对BSA和MC-BSA进行分子量测定,测定的BSA和MC-BSA相对分子量值分别是 66 455.70、76 515.84(图4)。MC-BSA分子量比BSA大,证实了MC-LR与BSA偶联成功,计算得到的MC-LR和BSA的偶联比为10 ∶1。
由于KLH没有精确的分子量,无法用质谱法准确测定MC-KLH的分子量,因此未采用此方法进行鉴定。
2.4 免疫鼠血清效价以及敏感度鉴定
如图5所示,以大于阴性血清吸光度2.1倍计,3只Balb/c小鼠的抗血清效价在1 ∶8 000~1 ∶32 000之间。选择效价最高且抑制效果最好的1号鼠抗血清用方阵滴定法优化包被原和抗血清工作浓度,优化后的包被抗原工作浓度为2.5 μg/mL,抗体稀释倍数为1 ∶8 000。在优化条件下用间接竞争ELISA法测定抗血清对微囊藻毒素LR的敏感度(图6),计算得到的 MC-LR对鼠抗血清的抑制中浓度(I50)为0.53 μg/mL。结果表明,试验合成的免疫抗原MC-KLH成功刺激小鼠产生免疫应答,并产生了能够灵敏识别MC-LR的抗体,由此确证了免疫抗原合成成功。
3 讨论与结论
3.1 免疫抗原和包被抗原的合成方法
目前常用的包被抗原和免疫抗原的合成方法有很多,含羧基(—COOH)的半抗原一般采用碳二亚胺法、混合酸酐法、N-羟基丁二酰亚胺活性酯法等合成。含氨基(—NH2)的半抗原一般使用戊二醇法、卤代硝基苯法、二异氰酸酯法、重氮化法等。含羟基(—OH)的半抗原一般可用琥珀酸酐法、光气法、卤代羧基法等合成。含有巯基(—SH)、醛(—CHO)、酮(—CO—)结构的半抗原亦可利用相应的双功能团试剂与蛋白偶联合成[18]。早期,Metcalf等将MC-LR与2-巯基乙胺反应形成氨基乙硫醇-MC-LR,从而引入1个游离氨基,再通过戊二醛法将其与载体蛋白KLH偶联[19]。Zeck等则将阳离子化卵清蛋白(cOVA)进行巯基化修饰后,与MC-LR第7位氨基酸(MdhA)进行偶联 [20]。盛建武等则在MC-LR第7位氨基酸分子上引入1个游离氨基,形成中间产物(H2N-et MC-LR),再使用戊二醇法将其分别与BSA和OVA偶联[21]。以上方法虽然获得了灵敏度较高的抗体,但是须要对MC-LR进行复杂的化学修饰后再与载体蛋白偶联。
本研究利用MC-LR的第3位(D-β-Me-Asp)和第6位(D-Glu)氨基酸中的2个游离羧基,用EDC·HCl和NHS将其与载体蛋白进行直接偶联。该方法具有无需半抗原修饰的优点,大部分试验易于掌握,而且偶联反应条件温和。另外,本试验采用了KLH作为免疫原的载体蛋白,KLH分子量巨大(400~800 ku),1个KLH分子带有数百个伯氨基基团,可以用于MC-LR偶联,相比于BSA或OVA可以引发更强烈的免疫应答。从小鼠免疫的实际效果来看,本试验用 MC-KLH 免疫的小鼠最高效价达到1 ∶32 000倍,免疫应答效果良好。
3.2 免疫抗原和包被抗原的鑒定方法
紫外扫描光谱法和SDS-PAGE是国内试验室最常用的2种偶联物鉴定方法,它们具有仪器设备价格低以及操作简便等优点。但这2种方法也存在了一定的局限性,如紫外扫描光谱法是通过观察偶联物的紫外吸收谱图与载体蛋白和半抗原相比有没有偏移或叠加来推测是否成功,是较为粗略的判断方法。以本研究为例,MC-KLH和MC-BSA的紫外谱图虽与载体蛋白存在差异,但偶联物并没有出现明显的 238 nm 处的半抗原特征吸收峰,因此难以准确判断是否偶联成功。另外用紫外扫描光谱法来计算半抗原与载体蛋白偶联比,是通过计算摩尔吸光系数来换算,结果并不精确。
SDS-PAGE法是通过比较载体蛋白和偶联物电泳迁移率的方法来判断偶联物分子量是否增大。对于本试验可以明显观察到MC-BSA的电泳迁移率低于BSA,证明偶联物分子量增大。但是对于许多分子量更小的半抗原以及偶联比极低情况,由于载体蛋白分子量较大(通常几十到几百ku),偶联了几十到一两百u的半抗原后,用此方法就难以区分。另外,也要注意一些电荷异常和构象异常及由亚基构成的蛋白可能不适用于SDS-PAGE测定分子量,如果采用这些蛋白作为载体蛋白偶联,鉴定时应考虑其他方法。
近年来兴起的基质辅助激光解吸电离飞行时间质谱技术,其准确度远远高于目前常规应用的SDS-PAGE与高效凝胶色谱技术,目前可测定生物大分子的分子量高达600 ku。它是通过对蛋白质分子离子化后通过质荷比进行分析,更能够准确地获得蛋白质相对分子质量,从而能精确计算出半抗原与载体蛋白的偶联比。本试验采用了此种方法精确测定了MC-BSA和BSA的分子量,最终确定了偶联比。
另外,本试验用于制备免疫抗原的KLH由于没有准确的分子量,难以用SDS-PAGE和MALDI-TOF MS对其偶联结果进行鉴定,但用该免疫原对小鼠免疫后获得了高效价和灵敏度的抗血清,最终证明了免疫原MC-KLH合成成功。
本研究成功合成了MC-LR的免疫抗原和包被抗原,为其单克隆抗体的制备奠定了基础。试验还系统比较了紫外扫描光谱、SDS-PAGE、MALDI-TOF MS等3种鉴定方法的适用性和优缺点,为其他人工抗原的鉴定方法提供了有益的参考。
参考文献:
[1]闫 海,潘 纲,张明明,等. 微囊藻毒素的提取和提纯研究[J]. 环境科学学报,2004,24(2):355-359.
[2]Humpage A R,Falconer I R. Oral toxicity of the cyanobacterial toxin cylindrospermopsin in male Swiss albino mice:determination of no observed adverse effect level for deriving a drinking water guideline value[J]. Environmental Toxicology,2003,18(2):94-103.
[3]Blahova L,Marsalek O B,Sejnohova L,et al. The first occurrence of the cyanobacterial alkaloid toxin cylindrospermopsin in the Czech Republic as determined by immunochemical and LC/MS methods[J]. Toxicon,2009,53(5):519-524.
[4]Meng G M,Sun Y,Fu W Y,et al. Microcystin-LR induces cytoskeleton system reorganization through hyperphosphorylation of tau and HSP27 via PP2A inhibition and subsequent activation of the p38 MAPK signaling pathway in neuroendocrine (PC12) cells[J]. Toxicology,2011,290(2/3):218-229.
[5]劉 媛,Tuomas H,刘贤金,等. 基于磁珠和时间分辨荧光免疫分析的微囊藻毒素LR单链抗体筛选与鉴定[J]. 中国农业科学,2012,45(2):330-337.
[6]顾丽丽. ELISA试剂盒法测定水中LR型微囊藻毒素[J]. 化学分析计量,2013,22(1):97-99.
[7]Rapala J,Erkomaa K,Kukkonen J,et al. Detection of microcystins with protein phosphatase inhibition assay,high-performance liquid chromatography-UV detection and enzyme-linked immunosorbent assay:comparison of methods[J]. Analytica Chimica Acta,2002,466(2):213-231.
[8]Dahlmann J,Budakowski W R,Luckas B. Liquid chromatography-electrospray ionisation-mass spectrometry based method for the simultaneous determination of algal and cyanobacterial toxins in phytoplankton from marine waters and lakes followed by tentative structural elucidation of microcystins[J]. Journal of Chromatography A,2003,994(1/2):45-57.
[9]Spoof L,Vesterkvist P,Lindholm T,et al. Screening for cyanobacterial hepatotoxins,microcystins and nodularin in environmental water samples by reversed-phase liquid chromatography-electrospray ionisation mass spectrometry[J]. Journal of Chromatography A,2003,1020(1):105-119.
[10]Fischer W J,Garthwaite I,Miles C O,et al. Congener-independent immunoassay for microcystins and nodularins[J]. Environmental Science & Technology,2001,35(24):4849-4856.
[11]Tsutsumi T,Nagata S,Hasegawa A,et al. Immunoaffinity column as clean-up tool for determination of trace amounts of microcystins in tap water[J]. Food & Chemical Toxicology,2000,38(7):593-597. [HJ1.83mm]
[12]Aranda-Rodriguez R,Kubwabo C,Benoit F M. Extraction of 15 microcystins and nodularin using immunoaffinity columns[J]. Toxicon,2003,42(6):587-599.
[13]Ward C J,Beattie K A,Lee E Y,et al. Colorimetric protein phosphatase inhibition assay of laboratory strains and natural blooms of cyanobacteria:comparisons with high-performance liquid chromatographic analysis for microcystins[J]. Fems Microbiology Letters,1997,153(2):465-473.[HJ1.72mm]
[14]Heresztyn T,Nicholson B C. Determination of cyanobacterial hepatotoxins directly in water using a protein phosphatase inhibition assay[J]. Water Research,2001,35(13):3049-3056.
[15]Kiviranta J,Sivonen K,Niemela S I,et al. Detection of toxicity of cyanobacteria by Artemia salina bioassay[J]. Environmental Toxicology & Water Quality,1991,6(4):423-436.
[16]Mcelhiney J,Lawton L A. Detection of the cyanobacterial hepatotoxins microcystins[J]. Toxicology & Applied Pharmacology,2005,203(3):219-230.
[17]Yu F Y,Liu B H,Chou H N,et al. Development of a sensitive ELISA for the determination of microcystins in algae[J]. Journal of Agricultural & Food Chemistry,2002,50(15):4176-4182.
[18]周思祥,劉福成. 农药人工抗原的合成研究进展[J]. 农药,2005,44(8):337-341.
[19]Metcalf J S,Bell S G,Codd G A. Production of novel polyclonal antibodies against the cyanobacterial toxin microcystin-LR and their application for the detection and quantification of microcystins and nodularin[J]. Water Research,2000,34(10):2761-2769.
[20]Zeck A,Eikenberg A,Weller M G,et al. Highly sensitive immunoassay based on a monoclonal antibody specific for[4-arginine]microcystins[J]. Analytica Chimica Acta,2001,441(1):1-13.
[21]盛建武,何 苗,宋保栋,等. 微囊藻毒素-LR完全抗原的设计及制备[J]. 环境科学,2005,26(3):33-37.
