氯氟醚菌唑合成方法述评
2019-08-14张志刚张奎祚郑晓迪何喜红孔令乐
张志刚,张奎祚,郑晓迪,何喜红,孔令乐,林 楠
(海利尔药业集团股份有限公司,山东青岛 266109)
氯氟醚菌唑(mefentrifluconazole)是巴斯夫研发、上市的第1个新型异丙醇三唑类杀菌剂,填补了三唑类杀菌剂10余年无新品上市的空白。氯氟醚菌唑化学名称 (2RS)-2-[4-(4-氯苯氧基)-α,α,α-三氟-邻甲基] -1-(1H-1,2,4-三唑-1-基)丙-2-醇;商品名Revysol;CAS登录号为[1417782-03-6];分子式为C18H15ClF3N3O2;相对分子质量为397.8。其化学结构式见图1。

图1 氯氟醚菌唑结构式
与其他三唑类杀菌剂一样,氯氟醚菌唑亦为甾醇生物合成中C14-脱甲基化抑制剂。氯氟醚菌唑分子中独特的异丙醇基团,使其能够非常灵活地从游离态自由旋转与靶标结合成为结合态,抑制壳针孢菌的转移,减少病菌突变,延缓抗性的产生和发展。灵活多变的空间形态使得氯氟醚菌唑对多种抗性菌株始终保持高效,是优秀的抗性管理工具。
氯氟醚菌唑表现出优异的生物性能,其广谱、高效,具有内吸传导性,兼具保护、治疗、铲除作用,速效、持效。其还可防控许多难以防治的真菌性病害,如锈病及壳针孢菌引起的病害等。氯氟醚菌唑适用于大田作物、经济作物和特种作物等世界范围内的60多种作物,如玉米、谷物、大豆、水稻等大田作物,以及青椒、葡萄等经济作物,并将应用于草坪和观赏植物等。氯氟醚菌唑将帮助全球种植户提升作物活力,提高作物产量和品质。氯氟醚菌唑既可叶面喷雾,也能用于种子处理。相对于其他唑类杀菌剂,氯氟醚菌唑不仅活性更高,而且拥有较好的环境特性,其对哺乳动物、蜜蜂等毒性较低,安全性较高。
2016年起,巴斯夫陆续向欧盟、美国、加拿大、墨西哥、巴西等国家和地区申请登记氯氟醚菌唑。2018年,氯氟醚菌唑在韩国取得登记,并率先上市。
1 工艺路线
文献报道的氯氟醚菌唑的合成路线主要有3条,具体如下。
路线1:由4-氟-2-三氟甲基苯乙酮与4-氯苯酚在碱性条件下发生取代反应生成1-[4-(4-氯苯氧基)-2-三氟甲基苯基] 乙酮,再与三甲基锍碘化物或三甲基锍甲基硫酸盐反应环氧化反应,生成2-[4-(4-氯苯氧基)-2-三氟甲基苯基] -2-甲基环氧乙烷,与1,2,4-三氮唑在碱性条件下发生开环反应得到氯氟醚菌唑[1-5]。

路线2:由1-溴-4-氟-2-三氟甲基苯在碱性条件下与4-氯苯酚发生取代反应,生成1-溴-4-(4-氯苯氧基)-2-三氟甲基苯,在异丙基氯化镁或氯化锂作用下,与乙酰氯发生反应,生成1-[4-(4-氯苯氧基)-2-三氟甲基苯基] 乙酮,再与液溴发生取代反应,生成2-溴-1-[4-(4-氯苯氧基)-2-三氟甲基苯基] 乙酮,然后与1,2,4-三氮唑在氢化钠作用下发生取代反应,生成1-[4-(4-氯苯氧基)-2-三氟甲基苯基] -2-[1,2,4] -三唑-1-基乙酮,再与甲基溴化镁发生格氏反应,得到氯氟醚菌唑[1-5]。

路线3:由4-溴-2-三氟甲基碘苯与异丙基氯化镁生成格氏试剂,与乙酰氯反应生成4-溴-2-三氟甲基苯乙酮,再与三甲基锍碘化物反应生成2-(4-溴-2-三氟甲基苯基)-2-甲基环氧乙烷,与1,2,4-三氮唑在碱性条件下发生开环反应,生成1-(4-溴-2-三氟甲基苯基)-1-甲基-2-(1,2,4-三唑-1-基)乙醇,再与双(频哪醇合)二硼发生反应得到1-[2-氯-4-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂戊环)-2-基] -1-甲基-2-(1,2,4-三唑-1-基)乙醇,加入双氧水氧化得到3-三氟甲基-4-[1-甲基-1-羟基-2-(1,2,4-三唑-1-基)乙基] 苯酚,最后在碱性条件下与4-氯氟苯发生醚化反应,生成氯氟醚菌唑[6]。
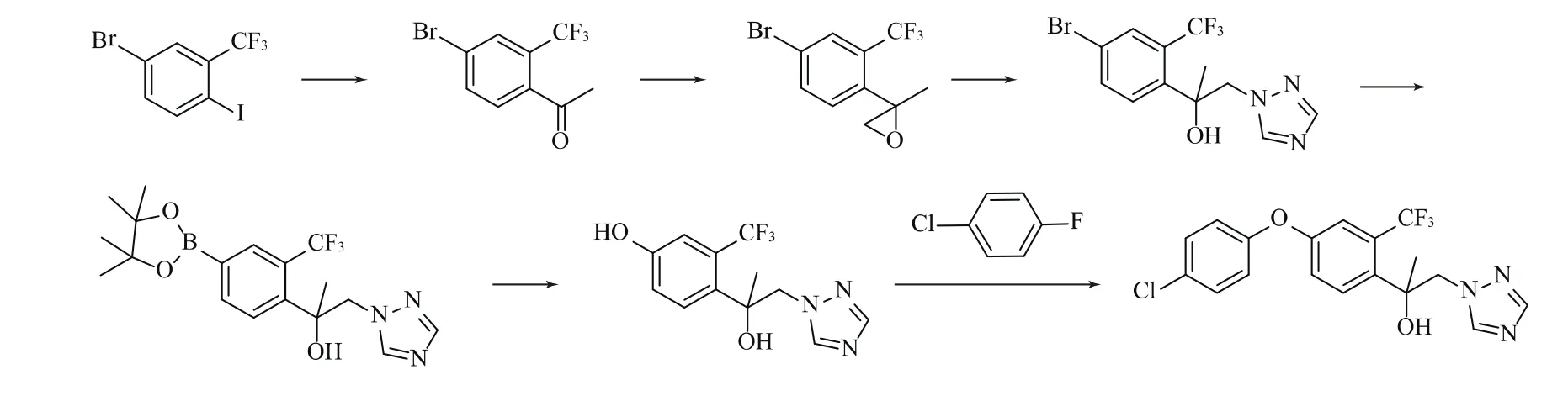
2 合成方法
2.1 合成方法1
将4-氟-2-三氟甲基苯乙酮622.0 g(3.02 mol)、4-氯苯酚426.7 g(3.32 mol)、碳酸钾542.1 g(3.92 mol)和DMF 2 365 mL混合,120℃下搅拌5 h,140℃下搅拌5 h。冷却后将该混合物加入盐水溶液中并用MTBE萃取3次。将有机相合并,用10%LiCl水溶液洗涤2次并干燥。蒸发溶剂得到中间体1-[4-(4-氯苯氧基)-2-三氟甲基苯基] 乙酮884.7 g,收率88%。
1H NMR(400 MHz,CDCl3)δ:2.60(s,3H)、6.98(d,2H)、7.10(d,1H)、7.30(s,1H)、7.35(d,2H)、7.50(d,1H)[1]。
将DMSO 140 mL(1.97 mol)加入氢化钠0.831 g(33 mmol)和THF 53 g的混合物中,冷却至约5℃。然后滴加DMSO 80 mL与三甲基锍碘化物6.42 g(31.5 mmol)的混合物,并将该混合物在5℃下搅拌1 h。然后滴加含中间体1-[4-(4-氯苯氧基)-2-三氟甲基苯基] 乙酮5.0 g(14.3 mol)的40 mL DMSO溶液,5 min内滴完。搅拌15 min,用饱和氯化铵溶液(150 mL)淬灭,并用MTBE萃取3次。将有机相合并,用水洗涤并干燥。蒸发溶剂,得到黄色油状物2-[4-(4-氯苯氧基)-2-三氟甲基苯基] -2-甲基环氧乙烷4.4 g,收率为89%。
1H NMR(400 MHz,CDCl3)δ:1.65(s,3H)、2.95-3.05(d,2H)、6.95(d,2H)、7.10(d,1H)、7.25(s,1H)、7.35(d,2H)、7.65(d,1H)[1,5]。
将2-[4-(4-氯苯氧基)-2-三氟甲基苯基] -2-甲基环氧乙烷1.92 g(4.96 mmol)、1,2,4-三氮唑1.715 g(24.8 mmol)、NaOH 0.496 g(12.41 mmol)和N-甲基吡咯烷酮48 mL的混合物在110℃下搅拌1 h,130℃下搅拌4 h。冷却至室温后,加入饱和氯化铵溶液,并将有机相用MTBE萃取3次。将有机相合并,用10%LiCl溶液洗涤2次并干燥。蒸发溶剂,加入二异丙醚后出现沉淀物,得到白色固体终产物2-[4-(4-氯苯氧基)-2-三氟甲基苯基] -1-[1,2,4] 三唑-1-基丙-2-醇1.55 g,收率为75%[1,5]。
2.2 合成方法2
使1-溴-4-氟-2-三氟甲基苯2.04 g与碳酸钾4.18 g在二甲基甲酰胺中混合,并将反应混合物加热至110℃。然后加入4-氯苯酚3.68 g(15.14 mmol),并将所得混合物在110℃下搅拌5 h。冷却,并用水/DCM萃取,将有机层用氯化锂水溶液洗涤,然后用氢氧化钠水溶液洗涤,干燥,过滤并蒸馏,得到油状物1-溴-4-(4-氯苯氧基)-2-三氟甲基苯3.14 g。
1H NMR(400 MHz,CDCl3)δ:6.80(d,1H)、6.95(d,2H)、7.35(d,2H)、7.55(d,1H)、7.80(s,1H)[1]。
在室温下,将1-溴-4-(4-氯苯氧基)-2-三氟甲基苯100.0 g(0.28 mol)溶于500 mL THF溶液中,再滴加异丙基氯化镁/氯化锂配合物284 mL(1.3 mol/L),搅拌2 h。将该混合物滴加到乙酰氯29.0 g(0.37 mmol)在500 mL THF中的溶液中。将所得反应混合物搅拌150 min,并用饱和氯化铵溶液淬灭。再用水/MTBE萃取后,将有机溶剂干燥并蒸发,得到黄色油状物1-[4-(4-氯苯氧基)-2-三氟甲基苯基] 乙酮96.6 g。
1H NMR(400 MHz,CDCl3)δ:2.6(s,3H)、7.0(d,2H)、7.10(d,1H)、7.30(s,1H)、7.37(d,2H)、7.50(d,1H)[1]。
将溴29.6 g(185 mmol)滴加到1-[4-(4-氯苯氧基)-2-三氟甲基苯基] 乙酮61.4 g(185 mmol)在700 mL乙醚溶液中,3 min内加完。将该混合物在室温搅拌90 min,然后在搅拌下缓慢加入1 L冰冷水和300 mL饱和碳酸氢钠溶液混合物,至pH值为7~8。将有机相用MTBE萃取2次,并用LiCl溶液洗涤。干燥并蒸发溶剂,得到褐色油状物2-溴-1-[4-(4-氯苯氧基)-2-三氟甲基苯基] 乙酮76 g,收率为83%。
1H NMR (400 MHz,CDCl3)δ:4.35 (s,2H)、7.0(d,2H)、7.12(d,1H)、7.34(s,1H)、7.38(d,2H)、7.55(d,1H)[1]。
将1,2,4-三唑3.76 g(53 mmol)缓慢分批加入氢化钠1.28 g(53 mmol)与150 mL THF混合物中,室温下搅拌30 min。向该溶液中滴加溶解在100 mL THF的2-溴-1-[4-(4-氯苯氧基)-2-三氟甲基苯基] 乙酮20.0 g(40.7 mmol),室温下搅拌约150 min。将反应混合物冷却至10℃,并缓慢滴加至冰冷水和氯化铵混合溶液中,将有机层用乙酸乙酯萃取3次。将有机相合并,干燥并蒸发溶剂。由二异丙醚重结晶得到白色固体中间体1-[4-(4-氯苯氧基)-2-三氟甲基苯基] -2-[1,2,4] 三唑-1-基-乙酮14.5 g,收率为84%。
1H NMR(400 MHz,CDCl3)δ:5.42(s,2H)、7.05(d,2H)、7.15(d,1H)、7.38(s,1H)、7.42(d,2H)、7.60(d,1H)、8.0(s,1H)、8.25(s,1H)[1]。
将2.4 mL LaCl3·2LiCl的THF溶液0.6 mol/L与1-[4-(4-氯苯氧基)-2-三氟甲基苯基] -2-[1,2,4] 三唑-1-基-乙酮0.5 g(1.31 mmol)溶于5.0 mL混合溶液中,并在室温下搅拌30 min。在室温下将所得溶液滴加到1-丙炔基溴化镁1.5 mL(0.5 mol/L)和THF混合溶液中,搅拌30 min,将所得混合物用10%HCl水溶液淬灭,并用MTBE萃取。有机相用盐水洗涤,干燥,蒸馏,在反相色谱柱上提纯之后,得到固体2-[4-(4-氯苯氧基)-2-三氟甲基苯基] -1-(1,2,4-三唑-1-基)戊-3-炔-2-醇25 mg,熔点为137℃。
按照类似方法,将1-[4-(4-氯苯氧基)-2-三氟甲基苯基] -2-[1,2,4] 三唑-1-基-乙酮的THF溶剂滴加到甲基溴化镁中,得到2-[4-(4-氯苯氧基)-2-三氟甲基苯基] -1-[1,2,4] 三唑-1-基丙-2-醇,熔点为121~122℃[1]。
2.3 合成方法3
3-三氟甲基-4-[1-甲基-1-羟基-2-(1,2,4-三唑-1-基)乙基] 苯酚的合成方法,参照WO2015/185708中类似物的合成方法,以4-溴-2-三氟甲基-1-碘苯为起始原料经过格氏反应,环氧化,开环,硼化和脱酯化5步反应得到[6]。
参照WO2015/185708中类似物的合成,以3-三氟甲基-4-[1-甲基-1-羟基-2-(1,2,4-三唑-1-基)乙基] 苯酚和4-氯氟苯为原料,在碱性条件下进行醚化反
3 小结
总结了氯氟醚菌唑经文献报道的3条合成路线,路线2和路线3路线相对较长,多步涉及到如格氏反应等危险工艺,产生“三废”量大,总收率相对较低。路线1所用原料易得,反应条件温和,路线短,收率高,合成成本低,是相对比较适宜的工业化路线。
