圈养不同性别林麝粪便菌群多样性研究
2019-08-08赵贵军朱吉彬郑程莉曾德军张承露戚文华封孝兰
赵贵军,竭 航,朱吉彬,郑程莉,曾德军,张承露,戚文华,封孝兰*
(1. 重庆市药物种植研究所,重庆 南川 408435;2. 四川养麝研究所,四川 成都 610016;3. 重庆三峡学院,重庆 万州 404000)
【研究意义】林麝 (MoschusberezovskiiFlerov)俗称獐子,是我国6种麝科动物中种群数量最多的一种。成年雄麝的香囊腺分泌物——麝香,不仅有较高的药用价值,还是一种名贵的天然高级香料。林麝为国家Ⅰ级重点保护野生动物,《世界自然保护联盟濒危物种红色名录》将林麝列为濒危物种[1-3]。从20 世纪60年代起我国便开始进行人工林麝养殖,但因环境恶化、疾病等因素规模仍旧难以扩大[4-5]。此外野生林麝自由采食多种植物的枝叶、果实和种子,一般不存在营养缺乏情况,但在人工饲养条件下,目前没有专门为林麝配制的全价饲料。青绿饲料品种单一,造成林麝肠道对营养吸收不全面,体质差,易患病[6]。随着微生态制剂的发展,利用有益菌去改善饲料配方,可提高林麝抗病性能[7-8]。【前人研究进展】健康宿主的肠道微生物为宿主提供营养和能量,有利于新陈代谢和免疫系统保持平衡。患病宿主的肠道微生物会引发系列炎症和增加导致疾病[9]。肠道微生物组成受到很多因素的影响,例如食性、健康状态、年龄、地理环境等。但对于不同性别健康林麝的肠道微生物的组成区别,对疾病抵抗力的差异,研究报道还较少。【本研究切入点】本研究拟通过性别差异下肠道微生物的组成特征,来反映不同性别差异下林麝的抗病能力,特别是揭示雄性林麝染病死亡率高的原因。【拟解决的关键问题】采用Illumina MiSeq测序技术,通过对不同性别的林麝粪便微生物组成的比较分析及肠道微生物功能的探究,了解不同性别下肠道微生物的差异对抗病性能及繁殖性能的影响。同时,揭示林麝的肠道微生物群落特征,为后续深入研究林麝的肠道消化吸收、疾病防控提供理论依据。
1 材料与方法
1.1 试验材料
试验动物为来自圈养于重庆市药物种植研究所的林麝,林麝饲粮以青绿饲料和精细饲料搭配饲喂。自由采食青绿饲料以红薯(Ipomoeabatatas)藤和光叶海桐(Pittosporumglabratum)嫩叶为主。精细饲料成分为玉米、大豆、小麦麸、菜籽饼和鱼粉,并添加磷酸氢钙、磷酸类、磷酸氢钙和食盐(表1)。试验共分2个处理组,分别为10只健康雄性林麝和10只健康雌性林麝,采集各林麝新鲜粪便5 g,用无菌聚乙烯自封袋装好锡箔纸包裹,快速置于液氮罐中带回实验室,然后于-80 ℃超低温冰箱中冻存以备DNA提取所用。
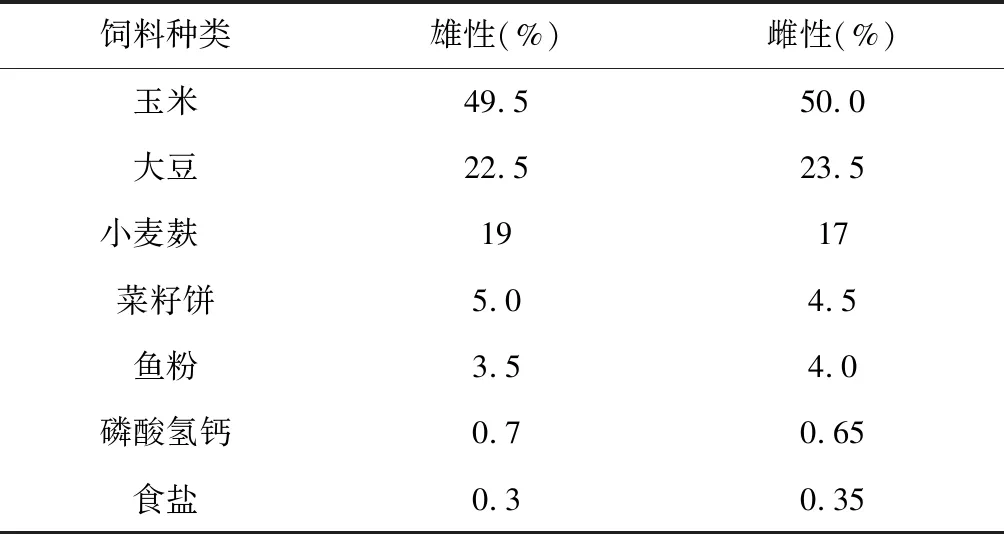
表1 林麝精细饲料组成
1.2 微生物总DNA提取
分别称取各粪便中心样品200 mg,放入2 mL离心管中,加入1.4 mL ASL缓冲液,振荡混匀后,用TIANamp Stool DNA粪便提取试剂盒(Tiangen Biotech, Beijing)提取林麝粪便中总DNA,具体操作步骤参照试剂盒说明书进行。通过0.8 %琼脂糖凝胶电泳检测DNA提取质量,同时采用Nano Drop 3300(Thermo Scientific)对DNA进行浓度检测。
1.3 目的片段PCR扩增及测序
选择16S rRNA基因的V3~V4区域作为扩增和测序的目的区间,扩增片段长度为480 bp。引物序列:338F:5′-ACTCCTACGGGAGGCAGCA-3′,806R:5′-GGACTACHVGGGTWTCTAAT-3′。PCR扩增采用NEB公司的Q5高保真DNA聚合酶,并严格控制扩增循环数,使循环数尽可能低的同时,也保证同一批样本的扩增条件一致。反 应 体 系25 μl:5×reaction buffer 5 μl,5×GC buffer 5 μl,dNTP(2.5 mM) 2 μl,Forwardprimer(10 μM) 1 μl,Reverseprimer(10 μM) 1 μl,DNA Template 2 μl,ddH2O 8.75 μl,Q5 DNA Polymerase 0.25 μl。PCR 扩增条件:98 ℃ 2 min;20~30个循环(循环数根据样品本身进行微调):98 ℃ 15 s,55 ℃ 30 s,72 ℃ 30 s,72 ℃ 5 min,10 ℃ 30 min。PCR扩增产物通过2 %琼脂糖凝胶电泳进行检测,并对目标片段进行切胶回收,回收采用AXYGEN公司的AxyPrepDNA 凝胶回收试剂盒。参照电泳初步定量结果,将PCR扩增回收产物进行荧光定量,荧光试剂为Quant-iT PicoGreen dsDNA Assay Kit,定量仪器为Microplate reader(BioTek,FLx800)。根据荧光定量结果,按照每个样本的测序量需求,对各样本按相应比例进行混合,采用Illumina公司的TruSeq Nano DNA LT Library Prep Kit制备测序文库。样本在派森诺生物公司进行测序,测序平台为IlluminaMiseq300PE。
1.4 测序数据分析方法
测序原始数据以FASTQ格式保存:R1. fastq和R2. fastq,Read 1和Read 2序列一一配对,采用滑动窗口法对FASTQ格式的双端序列逐一做质量筛查,随后,利用FLASH软件(v1.2.7,http://ccb.jhu.edu/software/FLASH/),对通过质量初筛的双端序列根据重叠碱基进行配对连接,将连接后的Barcode序列识别分配入对应样本,从而获得有效序列。接着运用QIIME软件[10](Quantitative Insights Into Microbial Ecology,v1.8.0,http://qiime.org/)识别疑问序列。除了要求序列长度≥150 bp,且不允许存在模糊碱基N之外,还将剔除:①5’端引物错配碱基数>1的序列;②含有连续相同碱基数>8的序列。随后,通过QIIME软件调用USEARCH(v5.2.236, http://www.drive5.com/usearch/)检查并剔除嵌合体序列。QIIME软件,调用UCLUST这一序列比对工具[11],对前述获得的序列按97 %的序列相似度进行归并和OTU划分,并选取每个OTU中丰度最高的序列作为该OTU的代表序列。对于每个OTU的代表序列分类地位鉴定,在QIIME软件中默认采用Greengenes数据库(Release 13.8,http://greengenes.secondgenome.com/),通过将OTU代表序列与对应数据库的模板序列相比对,获取每个OTU所对应的分类学信息。
获得各水平(界、门、纲、目、科、属)的注释文件后,使用R软件将门和科分类水平的鉴定结果绘制成柱状图,对OTU丰度矩阵中的全体样本在90 %的最低测序深度水平,统一进行随机重抽样,获得稀疏化(Rarefied)OTU丰度矩阵,绘制稀疏曲线和物种累积曲线。每个样本在相同测序深度下测得的Alpha多样性指数Chao1、ACE、Shannon、Simpson,使用IBM SPSS Statistics ver. 19进行显著性分析。通过R软件,对属水平的群落组成结构进行PCA分析,绘制二维图形,根据OTU丰度矩阵和样本分组数据构建PLS-DA判别模型。用QIIME软件进行ANOSIM分析,排列数设置为999,通过置换检验评价原始样本组间差异的统计学显著性。用PICRUSt(http://huttenhower.sph.harvard.edu/galaxy/tool_runner?tool_id=PICRUSt_normalize)[12]进行功能预测;基于京都基因组百科全书的KEGG orthology数据库,对同源基因功能进行归类。使用R软件,对丰度前50位的功能类群进行聚类分析并绘制热图。
2 结果与分析
2.1 测序质量分析
通过Illumina MiSeq测序平台,将测序所得原始双端测序拼接成一条序列,去掉低质量的序列及嵌合体序列最后获得有效序列。最终一共获得1 076 941条有效序列,雄性样本平均为(57 504±8187)条序列,雌性样本平均为(50 190±6963)条序列。按序列相似度大于97 %的标准对有效序列进行OTU聚类,最终共获得8292个OTUs,OTU个数雄性样本平均为2015±166,雌性样本平均为1801±385。
2.2 菌群的α多样性分析
根据观察雄性和雌性的OTUs的稀释曲线,0~1万多条序列测序深度,曲线急剧上升,不断发现新的OTUs,此后稀释曲线逐渐趋于缓慢,到达2万多条序列的测序深度时,测序的结果已足够反映当前样本所包含的多样性(图1A)。物种累积曲线在5~10个样本量时,呈现急剧上升的形态,不断发现大量的新物种,到达20个样本量时,曲线趋于平缓,表明样本量已足以反映群落的丰富度(图1B)。将所有样品标准化到 20 194条序列,得到4个多样性指数。经统计学t检验分析后发现,2组间无论是微生物丰富度还是微生物多样性在组间均无显著差异(P>0.05)。发现雄性的chao1、ACE、Shannon和Simpson 4个指标都高于雌性(表2),表明雄性微生物的丰富度多样性高于雌性。
2.3 不同分类水平菌群的组成差异
在本次研究中检测到的雄性肠道微生物共包含1个界(细菌),16个门,29个纲,46个目,87个科,156个属和176个种(表3);雌性肠道微生物共包含1个界(细菌),15个门,28个纲,48个目,89个科,155个属和176个种(表3)。可以发现雄性定义了更多的门,纲和属的细菌分类,而雌性定义了更多的目和科的细菌分类,同时雌雄2组定义了相同数目的种。

表2 林麝不同性别下粪便菌群多样性指数统计
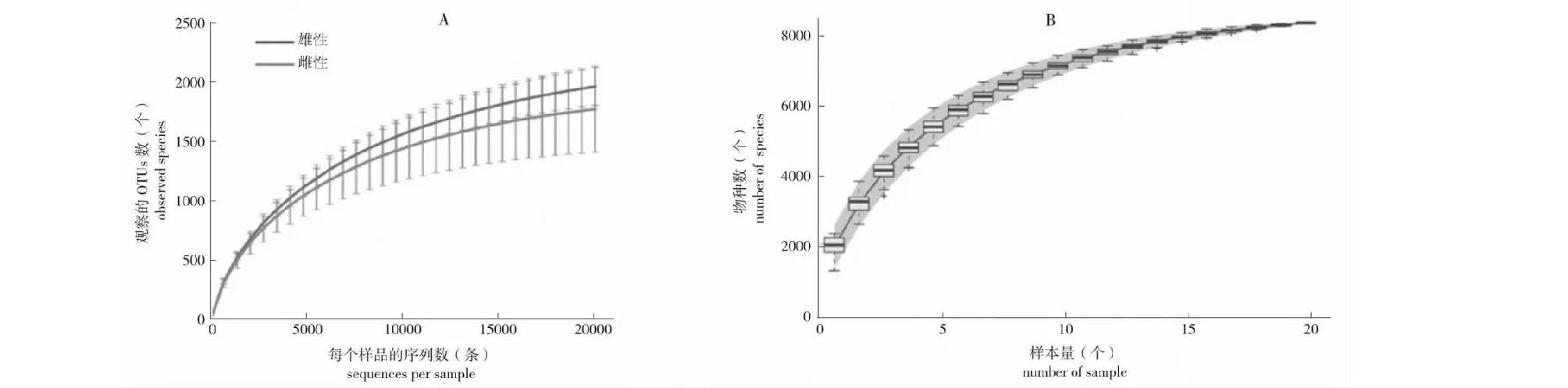
A:(观察的OTUs)的稀释曲线 ;B:物种累积曲线A:The rarefaction curve of observed_species; B:Species accumulation curves图1 林麝粪便菌群Alpha多样性分析Fig.1 Alpha diversity analysis

表3 林麝粪便20个样本在不同分类学水平上的群落组成
2.4 门水平菌群的组成差异
对雄性和雌性共有的17个主要门类进行了柱状图展示,其中在雄性主要的3个门类分别是Firmicutes(63.39 % ± 19.20 %),Proteobacteria(19.52 % ± 21.72 %)和Bacteroidetes(14.81 % ± 6.82 %),在雌性主要的3个门类分别是Firmicutes (57.27 % ± 12.57 %),Bacteroidetes(19.92 % ± 6.50 %)和Proteobacteria(18.44 % ± 16.12 %)。此外还有一些所占比例较小的门类,Tenericutes在雄性所占比例为0.87 % ± 0.63 %,在雌性占0.96 % ± 0.80 %。Verrucomicrobia在雄性所占比例为0.48 % ± 0.77 %,在雌性占1.59 % ± 2.83 %。发现不同的门类在不同性别中存在比例上的差异,雌性和雄性的第一大优势门类都是Firmicutes,而在第二大优势门类则不同,雄性是Proteobacteria,雌性是Bacteroidetes (图2 A)。在个体样本中,也发现比较明显的个体门类组成差异,雄性个体编号4209、4205、4199和4245这4个样本中Proteobacteria所占比例高,而其他雄性个体编号Proteobacteria所占比例低,同时雌性也存在个体上的组成差异现象(图2B)。
2.5 科水平菌落的组成差异
对雄性和雌性共有21个主要的科水平细菌分类进行了柱状图展示,雄性的主要科为Ruminococcaceae(26.73 % ± 10.35 %)、Lachnospiraceae(20.58 % ± 10.79 %)、*Clostridiales(11.88 % ± 4.80 %)、Pseudomonadaceae(10.80 %±17.52 %)、Bacteroidaceae(7.36 %±5.27 %) 和Enterobacteriaceae(7.16 %±13.43 %),雌性的主要科为Ruminococcaceae(24.71 % ± 6.04 %)、Lachnospiraceae(14.27 % ± 6.74 %)、*Clostridiales(12.72 % ± 5.01 %)、*Bacteroidales(7.89 % ± 6.60 %)Pseudomonadaceae(7.71 % ± 10.43 %)和Bacteroidaceae(6.32 % ± 4.36 %)。Ruminococcaceae,Lachnospiraceae和*Clostridiales都是雌雄在科水平的主要优势菌群,此外雄性的第4大优势菌群是Pseudomonadaceae,而雌性的第4大优势菌群是*Bacteroidales,Enterobacteriaceae在雄性所占比例高于雌性。S24-7所占比例较低,在雌雄中存在显著差异,在雄性中含量更高。结果可以明显反映出雌雄的科水平的门类组成比例差异(图2 C)。在个体样本中,发现雄性样本4199的第一大优势菌群是Enterobacteriaceae;Moraxellaceae在雌性样本5010中所占比例较高;Verrucomicrobiaceae在雌性样本4244中所占比例较高。相同细菌在不同个体所占比例的差异直接反映了个体差异的存在(图2D)。
2.6 聚类差异比较分析
对2个试验组共计20个样本进行主成分分析及以偏最小二乘回归的判别模型分析。主成分分析中PC1的贡献值为41.08 %,部分雄性和雌性的距离非常近,雄性更多地分布在左边区域;PC2的贡献值为23.43 %,也存在雌性和雄性相似的情况,但雌性更多地分布在下方区域。发现雌性和雄性也存在一定的差异,但是聚类的效果不太好,主要是由个体差异造成的(图3 A)。为了更好展示雌性和雄性之间存在的差异,进行了以偏最小二乘回归的判别模型分析,发现雌性和雄性两组之间的样本距离比较远,明显反映出两者之间存在一定的差异,不过在同一分组中雌性样本距离分散,重复性不好,而在同一分组中雄性样本距离近,重复性好(图3 B)。此外,在考虑到物种存在情况下未加权的UniFrac(ANOSIM:R2= 0.020,P=0.025)存在显著差异,也可以反映出雌雄之间存在一定的差异性。
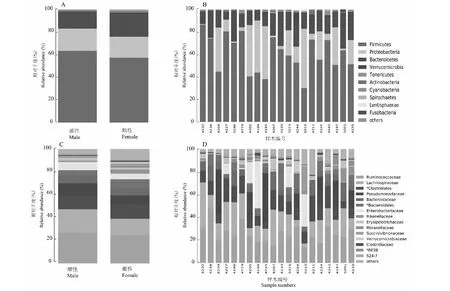
A. 雌性和雄性主要的门水平组成,B. 20个样本的主要门水平组成,C. 雌性和雄性主要的科水平组成,D. 20个样本的主要科水平组成;每个条带代表一个分组细菌平均的相对丰度,或代表一个个体细菌的相对丰度。门水平排名前10和属水平排名前15的细菌分类被展示。others代表其他细菌分类及未注释出来的细菌分类,若科水平菌群没有菌名或菌被注释为others,则用其科以上菌名加“*”表示A:Microbial composition of different groups at Phylum level; B:Microbial composition of 20 individuals at Phylum level; C:Microbial composition of different groups at family level; D:Microbial composition of 20 individuals at family level;Each bar represents the average relative abundance of each bacterial taxon within a group or individuals. The top 10 abundant taxa are shown at Phylum level and 15 abundant taxa are shown at family level.Others represent other bacterial taxa and undefined bacterial taxa.Moreover,sequences without bacterial name or assigned as others would be defined at family level marked with‘*’图2 林麝粪便不同分组和个体的微生物组成Fig.2 Microbial composition of different groups and individuals
2.7 菌群代谢功能聚类分析
在丰度前50的功能类群聚类的热图(图4,封三)发现一定的功能差异,可以看到明显的红色和绿色的模块分区。特别是4199、4232、4227、4196和4207这几个雄性样本存在很多相同的功能类群,其他的雄性样本存在一定模块区域相似性,也反映出来了一定的个体差异。通过菌落功能类群聚类发现雌性和雄性微生物代谢功能存在差异,为了更加准确地反映这种差异性,还需要进行宏基因组研究。
3 讨 论
本研究中主要利用Illumina MiSeq测序平台进行高通量测序研究了人工饲喂下雄性和雌性林麝微生物肠道微生物的组成。结果显示,人工饲喂下雄性和雌性肠道微生物的丰富度和多样性不存在显著差异,雄性的丰富度和多样性要略微高于雌性。之前,有学者对野生-圈养林麝及健康-腹泻林麝进行研究,也没有发现肠道微生物的丰富度和多样性存在显著性的差异[13],而在高山麝-林麝的研究中发现了显著差异[14],说明物种间的差异性高于物种内部之间的差异性。这与前人关于人类性别差异对肠道微生物丰富度和多样性影响的研究结论“女性的普遍高于男性”相反。当然背景差异也会影响,比如:年龄、饮食习惯、地理位置、健康状况等[13-14]。此外,在从肉食性到草食性的转变中,肠道细菌的多样性也会显著增加[15]。大部分研究认为,肠道微生物的丰度和多样性越高,越能反映出一个健康的肠道环境,维持微生态系统的动态平衡,确保正常的生理功能。
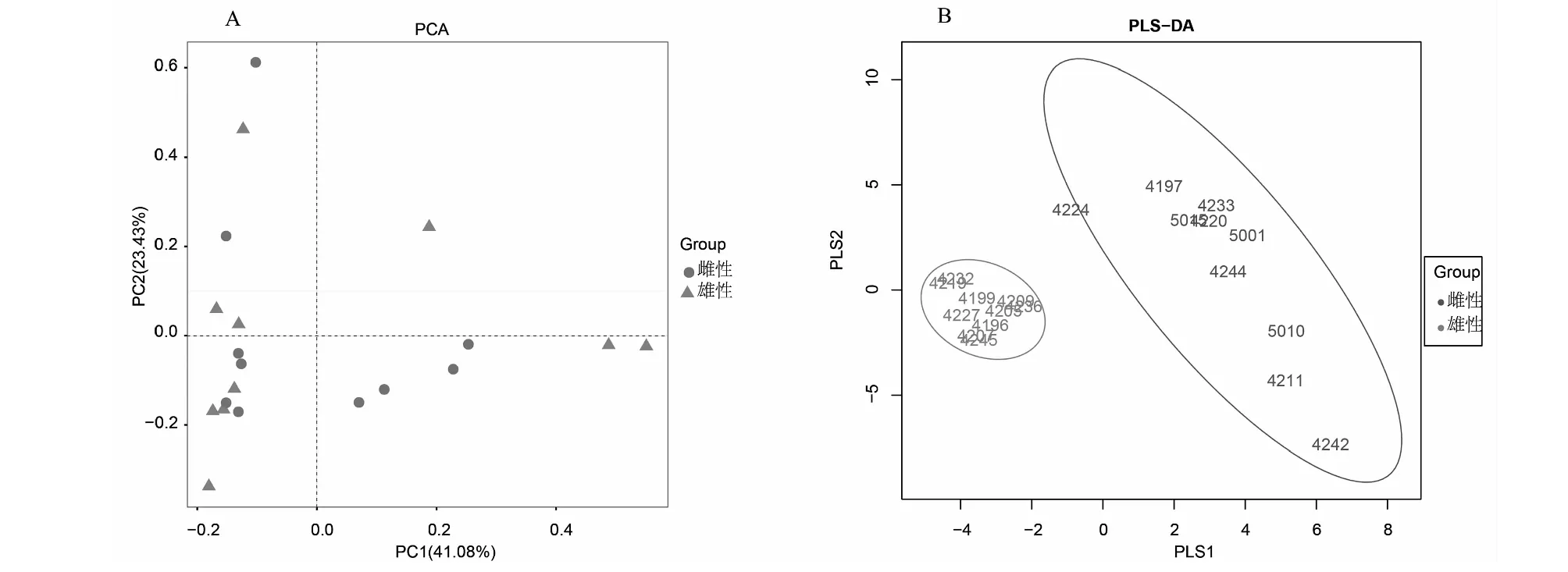
A. 基于欧式距离的三维主成分分析(PCA);B. 以偏最小二乘回归的判别模型(PLS-DA)A:Three-dimensional plot of principal component analysis (PCA) based on Euclidean distance;B:Discriminant model with partial least squares regression (PLS-DA) 图3 林麝粪便微生物丰度矩阵聚类图Fig.3 Abundance matrix clustering
相对丰度的柱状图显示了无论雌性还是雄性主要的优势菌都是厚壁菌门(Firmicutes),雄性林麝所占比例为63.39 %±19.20 %,雌性林麝所占比例为57.27 %±12.57 %。本文的研究结论基本上与之前对食草动物肠道细菌群落的描述是一致的,尤其是反刍动物肠道微生物群[16-18]。厚壁菌门组成的细菌类型,能够分解食物中的纤维素并将纤维素降解为宿主的挥发性脂肪酸,还可以调节体内的免疫反应,抑制机会性病原体的入侵并预防肠道炎症。其中瘤胃菌科(Ruminococcaceae),毛螺旋菌科(Lachnospiraceae)和梭菌目(*Clostridiales)都属于厚壁菌门。拟杆菌门(Bacteroidetes)细菌的主要功能是帮助宿主降解碳水化合物(尤其是多糖)、蛋白质和其他物质,以提高宿主的营养利用率[19],雄性林麝所占比例为14.81 %±6.82 %,雌性林麝所占比例为19.92 %±6.50 %,由此可见拟杆菌门更多富集在雌性肠道中。拟杆菌科(Bacteroidaceae)和拟杆菌目(*Bacteroidales)为拟杆菌门中占优势细菌分类。这两种细菌门类组合也是典型瘤胃微生物区系,帮助宿主消化动物体自身难以利用的粗纤维,从大量的低营养的植物纤维中摄取所需的能量,而且参与分解代谢有毒物质[20]。
本研究中发现变形菌门(Proteobacteria)在雄性林麝所占比例为19.52 %±21.72 %,雌性林麝所占比例为18.44 %±16.12 %,两组不存在显著差异,变形菌门在雄性林麝所占比例高于雌性林麝,并且在雄性中个体差异比较大。之前周美丽等[21]在对6只健康圈养雄性林麝研究中,发现变形菌门所占比例为5.2 %±5.6 %,也发现相同的细菌分类在不同个体中存在明显的差异,但是本文变形菌门所占的比例是之前研究的3倍之多。Mao等人[22]在奶牛的小肠中发现了大量变形菌门,在粘膜样本中所占比例为37.26 %,消化物样本中高达45.6 %。因此,在不同的动物个体、不同肠道部位和样本来源中变形菌门的定植也许是可变的。变形菌门的流行率是不稳定微生物群落的一个标志(生态失调)和疾病的潜在诊断标准,低纤维饮食和急性或慢性炎症都会导致变形菌门在胃肠道富集[23]。可以推测,在该研究中人工圈养的林麝健康状况不佳,可能存在慢性炎症的情况。假单胞菌科(Pseudomonadaceae)和肠杆菌科(Enterobacteriaceae) 属于变形菌门,这两类细菌对宿主具有潜在致病的机会,本文研究中在雄性林麝所占比例高于雌性林麝,而且在之前Li等人[12]对腹泻与健康林麝的肠道微生物研究中发现,腹泻组包含有更多肠杆菌科下的细菌属类,间接证明了该研究中雄性林麝更易于患上腹泻等疾病。
在主成分和回归的判别模型及菌群代谢功能的聚类分析中,雄性和雌性在细菌聚类距离和功能作用上存在一定的差异性。与雌性相比之下雄性10个样本距离近,重复性效果好。Li等人[12]对野生和圈养林麝的肠道微生物研究中,在圈养中发现更多的变形菌门。野生林麝以各种高纤维植物的叶子为食,圈养的林麝以人工喂养的树叶、高蛋白和多糖为食,而这两种喂养方式之间存在着巨大的差异。Filippo等人[23]在非洲与欧洲儿童研究中,非洲儿童主要富集更多的拟杆菌门,欧洲儿童富集更多的厚壁菌门。而饮食习惯在其中扮演着重要的角色,非洲儿童是典型的素食者,食物组成里脂肪和动物蛋白含量较低,主要是富含淀粉、纤维和植物多糖的食物,而欧洲儿童是典型的西方饮食,富含动物蛋白、糖、淀粉、脂肪和低纤维;非洲儿童的肠道细菌产生更多的短链脂肪酸有利于抵御疾病,而欧洲儿童含有更多Enterobacteriaceae等潜在致病菌,更容易发生肠道疾病。对比之下,在雄性中发现更多的厚壁菌门和变形菌门,而在雌性中发现更多的拟杆菌门。可以推测,雄性更偏好饲喂的精细饲料,摄入的食物成分相似造成微生物组成差异小,而部分雌性林麝更偏好自由采食的青绿饲料,所以造成雄性和雌性环境适应能力及抗病性能差异,但是这种雌雄差异存在很大的个体差异性。目前,圈养的林麝总是有很高的肠道疾病发病率,也反映出目前的精细饲料并不能完全满足林麝的营养需求,需提高青绿饲料的比重和多样性,模拟野外林麝摄食组成,更有利于林麝的健康成长。
4 结 论
通过高通量测序和生物信息学分析对林麝的粪便微生物群进行了研究,结果显示,林麝的粪便微生物以厚壁菌门、拟杆菌门和变形菌门为主。雄性林麝和雌性林麝在微生物的丰富度和多样性不存在显著差异,在微生物的组成成分上相似,不同细菌分类在个体上有明显的变化。细菌功能聚类分析反映了雌性和雄性存在一定差异,是饮食偏好差异还是宿主基因型差异造成的还需要进一步研究。该研究圈养下的林麝肠道微生物含有较高比重的变形菌门,存在很多机会致病菌。建议应增加饲料中的粗纤维成分,防止林麝出现严重的肠道疾病。随着高通量技术的发展,不仅是林麝粪便微生物的研究,应该全面展开对健康与非健康不同肠段林麝肠道微生物的研究,将直接有利于林麝养殖业的发展。
