应用高通量测序技术分析大菱鲆幼鱼肠道及其养殖环境的微生物群落结构*
2019-08-05吴欢欢王伟继胡玉龙
吴欢欢 王伟继 吕 丁 胡玉龙 孔 杰
应用高通量测序技术分析大菱鲆幼鱼肠道及其养殖环境的微生物群落结构*
吴欢欢1,2王伟继2吕 丁2胡玉龙2孔 杰1,2①
(1. 上海海洋大学水产与生命学院 水产种质资源发掘与利用教育部重点实验室 上海 201306; 2. 中国水产科学研究院黄海水产研究所 农业农村部海洋渔业资源可持续利用重点实验室 青岛 266071)
采用基于Illumina测序平台的高通量测序技术,对大菱鲆()幼鱼肠道及其养殖水体、生物饵料中细菌种类及丰度进行研究。测序结果显示,养殖水体、生物饵料和大菱鲆幼鱼肠道等19个样品共获得有效序列547621条,可聚类于3771个可分类操作单元(OTUs),归属于养殖水体、生物饵料、健康幼鱼和发病幼鱼的操作分类单元(OTU)个数分别为3038、1090、87和777,其中,健康幼鱼与生物饵料、健康幼鱼与养殖水体特有的OTU个数分别为57和0,发病幼鱼与生物饵料、发病幼鱼与养殖水体特有的OTU个数分别为481和31。表明幼鱼肠道微生物多样性与生物饵料密切相关。根据细菌注释结果,拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)和变形菌门(Proteobacteria)在大菱鲆幼鱼肠道中占优势地位,其中,健康幼鱼肠道微生物共聚类为8个门,发病幼鱼的肠道微生物可聚类为19个门。与健康幼鱼相比,发病幼鱼肠道门水平上的3种主要优势菌群落结构出现失衡。此外,对各样品中丰度最高的100位OTU分析显示,幼鱼肠道优势菌种类与生物饵料中的优势菌种类密切相关,而每个发病幼鱼肠道优势菌种类具有一定的独立性。本研究旨在为大菱鲆健康养殖和微生态调控提供实验依据。
生物多样性;高通量测序;大菱鲆
大菱鲆()属于比目鱼类,在我国市场上称为“多宝鱼”。野生大菱鲆生活于冰岛与摩洛哥附近的欧洲海域(Blanquer, 1992),是一种具有耐低温、生长快等特点的大型底栖经济鱼类。近年来,由于缺乏抗病大菱鲆良种选育,腹水症成为苗种培育期间的多发病,对大菱鲆养殖业造成了严重影响。腹水症病原学的初步研究认为,细菌是导致腹水症发生的主要原因(景亚运等, 2016)。但多年来,研究人员对大菱鲆腹水症的致病菌的研究表明,导致大菱鲆发生腹水症的致病菌却不尽相同,如迟缓爱德华氏菌()(李筠等, 2016)、鳗弧菌()(张晓君等, 2006)、灿烂弧菌()(Thomson, 2005)、哈维氏弧菌()(Austin, 2006)和溶藻弧菌()(张伟妮, 2006)等,腹水症这一特征给该病的诊断和防治造成了困难。有学者利用限制性片段长度多态性分析(RFLP)研究大菱鲆肠道微生物多样性(邢孟欣等, 2014),但这种传统方法获得的生物信息较少,具有一定的局限性,只能对少数主要的优势微生物进行分析(王贤丰等, 2017),难以全面解析微生物的组成结构。
高通量测序技术是一种能够对上百万个脱氧核苷酸链同时进行测序的第2代技术。目前,基于细菌16S rDNA扩增子测序的高通量测序技术成为研究样本生境内环境微生物菌群结构的重要手段,在水环境(窦妍等, 2016)、土壤(Ligi, 2014; Zhang, 2016)、空气(李红梅等, 2015)、口腔(Pozhitkov, 2011)和植物生境(陈泽斌等, 2016)等方向得到了广泛应用,全面解析样品生境内的微生物组成和数量。
本研究基于Illumina测序平台的高通量测序技术对山东某大菱鲆养殖场育苗期的养殖水环境、蓄水池中自然海水、生物开口饵料和健康与自然患腹水症的幼鱼样品进行研究,分析暴发腹水症期间投喂的生物饵料、养殖水体和幼鱼肠道的生物环境内的微生物组成及数量变化,以期更深入了解大菱鲆育苗肠道及养殖环境群落结构,为大菱鲆健康养殖和微生态调控提供实验依据。
1 材料与方法
1.1 实验材料
于2017年4月下旬在山东某养殖场应用工厂化海水半循环养殖系统进行大菱鲆家系苗种培育。苗种培育初期(5月下旬)育苗车间开始流行腹水症,期间收集了幼鱼(Group_Fish)、生物饵料(Group_ Baits)和养殖水体(Group_Water) 3类样品。幼鱼分为健康幼鱼组(Group_E)和发病幼鱼组(Group_F),每组3尾;生物饵料有来自渤海湾的金海湾小卤虫()和无棣小卤虫()、来自西藏的大卤虫()、轮虫()、小球藻()和裂壶藻(),每种饵料1个样品;养殖水体可分为蓄水池自然海水(Group_B)、健康幼鱼养殖水体(Group_C)和发病幼鱼养殖水体(Group_D),其中,自然海水1个,养殖水体各3个。
1.2 实验方法
1.2.1 样品收集 生物饵料中收集的卤虫和轮虫均由卵孵化去壳收集得到;小球藻和裂壶藻为公司培养收集得到;水样是利用隔膜真空泵抽滤2 L水体过0.22 μm孔径的醋酸纤维滤膜得到的。
1.2.2 DNA的提取 水环境DNA使用天根土壤DNA提取试剂盒(TIANamp Soil DNA Kit)提取,具体操作参照试剂盒说明书;其余样品DNA采用酚–氯仿法提取。
1.2.3 高通量测序方法 利用细菌通用引物343F: 5'-TACGGRAGGCAGCAG-3'/798R: 5'-CCGTCAATTCMTTTRAGTTT-3'对细菌的16S rDNA的V3-V4区进行PCR扩增,送诺和致源生物公司进行基于Illumina平台的高通量测序。
1.2.4 生信分析 对下机后的原始数据(Raw reads)进行双端去杂、拼接和去嵌合体等质控处理,获得优质的有效序列(Valid tags)。应用VSEARCH(v2.4.2)软件,对Valid tags依照97%的相似度聚类成为可操作分类单元(Operational taxonomic unit, OTU),而后选取每个归类单元中丰度最高的序列作为该单元的代表序列(Edgar, 2010),并与Silva的SSU rRNA数据库序列信息比对,获得生物注释。最后对各样品OTU进行丰度、α-多样性、β-多样性以及各分类水平上的细菌群落结构比较分析,来探究饵料和养殖水体对大菱鲆幼苗生物多样性的影响。
2 结果与分析
2.1 高通量数据统计
19个样品所测的原始序列经去杂、拼接等质控处理共得Clean tags 722213条,然后再去除嵌合体,共获得有效序列547621条,占Clean_tags的75.83%;使用VSEARCH (v2.4.2)软件,对有效序列按照97%的相似度共聚类为3771个OTUs。
生物饵料中海大小卤虫(B1)、无棣小卤虫(B2)、无棣大卤虫(B3)、轮虫(B4)、小球藻(B5)和裂壶藻(B6)分别获得的OTU个数为90、743、50、258、277和111,去重复后饵料组共得1090个OTUs。水体组共获得3080个OTUs。其中,自然海水的OTU个数为2124,3个检测的健康幼鱼养殖水体的OTU个数分别为1899、1806和1013,去重复后健康幼鱼养殖水体组共获得2464个OTUs,3个检测的发病幼鱼养殖水体的OTU个数分别为520、1374和1320,去重复后发病幼鱼养殖水体组共获得1560个OTUs。幼鱼组共获得785个OTUs。其中,3尾健康幼鱼的OTU个数分别为44、51和75,去重复后健康幼鱼组共获得87个OTUs,3尾发病幼鱼的OTU个数分别为219、634和41,去重复后发病幼鱼组共获得777个OTUs。根据各样品OTU信息,对各样品组进行统计如表1所示。
表1 各组OTU统计分析

Tab.1 Statistical analysis of OTUs in different groups
2.2 自然海水组、健康幼鱼组、发病幼鱼组和饵料组的OTU Venn图分析
OTU的数目可以代表物种的丰富程度(Zhang, 2009)。为探究养殖水体、饵料对幼鱼生境内生物多样性的影响程度,对自然海水组、健康幼鱼组、发病幼鱼组和饵料组的OTU进行Venn图分析(图1)。

图1 各组韦恩图
Group_B: 自然海水组; Group_E: 健康幼鱼组; Group_F: 发病幼鱼组; Group_Baits: 生物饵料组
Group_B: Natural sea water; Group_E: Healthy juvenile fish; Group_F: Diseased juvenile fish; Group_Baits: Living baits
Group_E和Group_F: Group_E(健康幼鱼组)和Group_F(发病幼鱼组)的OTU个数分别为87和777,共有的OTU个数为79个,分别占各自组OTU数目的90.80%和10.17%。
Group_Water: 自然海水组(Group_B)的OTU个数为2124,健康养殖水体组(Group_C)的OTU个数为2464,发病养殖水体组(Group_D)的OTU数有所降低,为1560,Group_Water总OTU数为3038个。其中,健康养殖水体组(Group_C)与自然海水组(Group_B)共有的OTU数为1702个,占自然海水组总数的80.13%;而发病养殖水体组与自然海水组共有的OTU个数为681个,占自然海水组OTU总数的32.06%。
Group_E、Group_B和Group_Baits: 3组共有的OTU数目为23个。其中,Group_E与Group_Baits共有的OTU数为80个,特有的OTU数为57个,分别占Group_E OTU数的91.95%和65.52%;Group_E与Group_B共有的OTU数为23个,特有的OTU数为0,分别占Group_E OTU数的26.44%和0。
Group_F、Group_B和Group_Baits: 3组共有的OTU数目为155个。其中,Group_F与Group_Baits共有的OTU数为636个,特有的OTU为481个,分别占Group_F OTU数的81.85%和61.90%;Group_F与Group_B共有的OTU数为186个,特有的OTU为31个,分别占Group_E OTU数的23.94%和3.99%。
2.3 微生物多样性分析
2.3.1 生境内多样性(α-Diversity)分析 使用软件Qiime计算各样本均一化处理后OTU数据的Goods coverages、Chao1和Simpson指数(表2),分别表征数据的测序深度、OTU丰富度和群落多样性。所有样品的Good’s coverages指数范围为0.93~1.00。基于Chao1指数绘制的稀释曲线如图2所示,随着抽样数的增加,各样品稀释曲线斜率均逐渐降低,后期曲线趋于平缓,Chao1指数基本与测序获得的OTU数目一致,说明在当前的测序深度下,得到的测序数据量可以代表样本中的细菌信息。Group_Baits、Group_Water和Group_Fish的Simpson指数分别为0.83±0.094、0.95±0.035和0.82±0.12,利用SPSS 18.0软件对Simpson指数进行单因素方差分析(One-way ANOVA),显示Group_Water的微生物多样性程度均显著高于Group_Baits和Group_Fish两组(<0.05)。
2.3.2 生境间多样性(β-Diversity)分析 微生物群落聚类分析根据样本相似性进行非加权组平均法(Unweighted pair group method with arithmetic mean, UPGMA)分析,采用Jackknifed重复抽样对UPGMA的可靠性进行检验(图3)。结果显示,75%~ 100%可信度认为水体组中的各样本单独聚为一支;50%~75%可信度认为饵料组中B2、B4和B5与发病幼鱼组中F15.1和F15.2聚为一支;75%~100%可信度认为饵料组中的B1、B3和B6、发病幼鱼组中F15.3与健康幼鱼组所有样品聚为一支。
表2 各样品α多样性指数

Tab.2 α-Diversity indices of each sample
利用主成分分析(PCA, Principal component analysis)对反应样本差异的OTU组成进行方差剖分,降维成2个反映方差的2个特征值,并以此作为二维坐标PCA图的横纵坐标,基于Unifrac距离进行PCA分析。从图4可明显看出,所有样品大致聚为5簇:饵料与鱼体组(Group_Baits、Group_E and Group_F) 2簇,自然海水组(Group_B)、健康养殖水体组(Group_C)、发病养殖水体组(Group_D)各一簇。Group_Baits、Group_C和Group_D大部分样品均重叠在一簇。
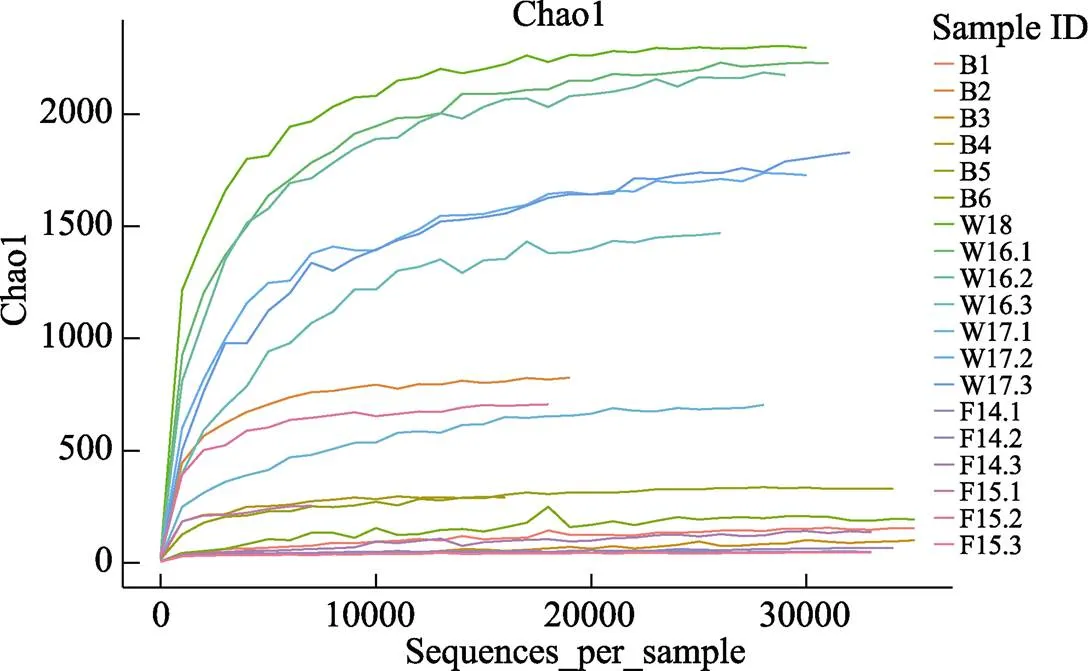
图2 基于Chao1指数的稀释曲线
B1: 金海湾小鲁虫; B2: 无棣小鲁虫; B3: 西藏大鲁虫; B4: 轮虫; B5: 小球藻; B6: 裂弧藻; W18: 自然海水; W16: 健康幼鱼养殖水体; W17: 发病幼鱼养殖水体; F14: 健康幼鱼; F15: 发病幼鱼。同图3、图5和图6
B1:in Jinhaiwan; B2:in Wudi; B3:in Tibet; B4:; B5:; B6:; W18: Sea water; W16: Aquaculture water of healthy juvenile fish; W17: Aquaculture water of diseased juvenile fish; F14: Healthy juvenile fish; F15: Diseased juvenile fish. The same as in Fig.3, Fig.5, and Fig.6

图3 UPGMA聚类分析及可信度检验
红色、黄色、绿色、蓝色分别代表UPGMA分析可信度的75%~100%、50%~75%、25%~50%和25%
Red, yellow, green, and blue represent 75%~100%, 50%~75%, 25%~50%和25% of UPGMA analysis reliability
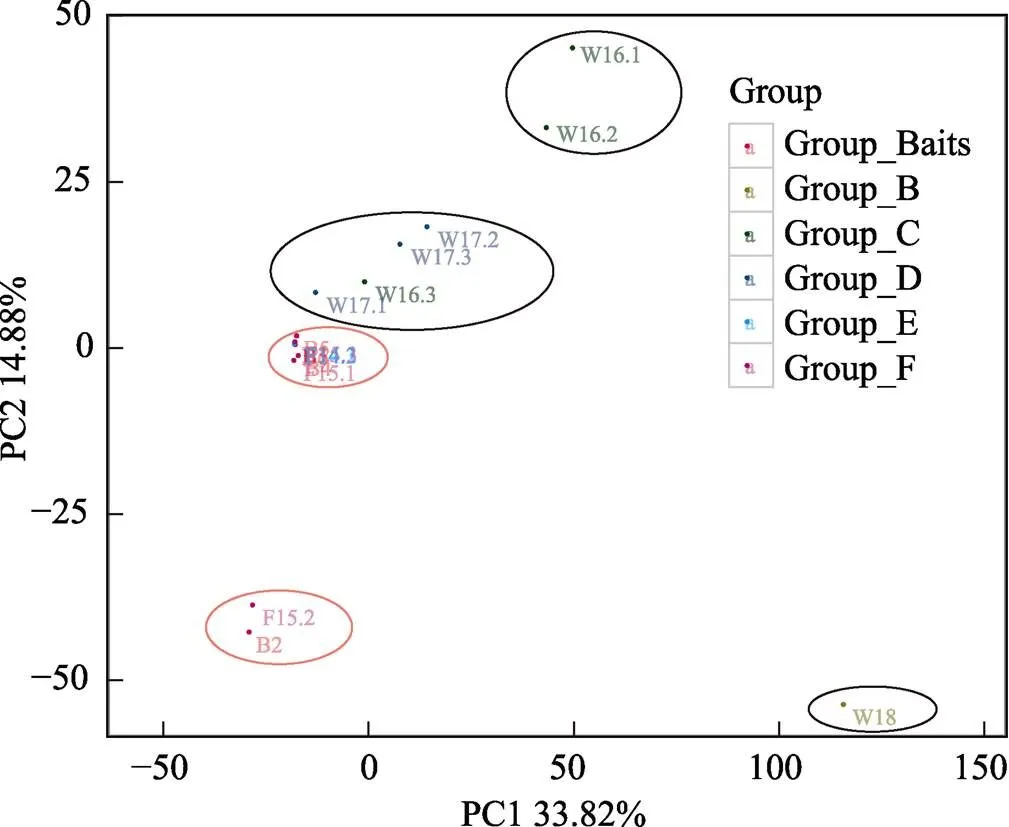
图4 主成分分析
Group_B: 自然海水组; Group_C: 健康幼鱼养殖水体; Group_D: 发病幼鱼养殖水体; Group_E: 健康幼鱼组; Group_F: 发病幼鱼组; Group_Baits: 生物饵料组
Group_B: Natural sea water; Group_C: Sea water of healthy juvenile fish; Group_D: Sea water of diseased juvenile fish; Group_E: Healthy juvenile fish; Group_F: Diseased juvenile fish; Group_Baits: Living baits
2.3.3 门、科、属三种分类水平下的群落结构分析
根据注释结果,所有样本在本次测序中检测的微生物有细菌和古细菌,其中,细菌归于37个门、100个纲、225个目、459个科和959个属。
在门水平上,纤维杆菌门(Fibrobacteres)、迷踪菌门(Elusimicrobia)、黏胶球菌门(Lentisphaerae)、绿弯菌门(Chloroflexi)、疣微菌门(Verrucomicrobia)、Latescibacteria、Cloacimonetes、Aminicenantes、装甲菌门(Armatimonadetes)、Saccharibacteria、衣原体门(Chlamydiae)、螺旋菌门(Spirochaetae)、Synergistetes、Candidate_division_SR1、Marinimicrobia_(SAR406_ clade)、TM6、PAUC34f、JL_ETNP_Z39、GOUTA4、SHA_109、WCHB1_60和TA06等22个门细菌含量较低,占比不足细菌总量的1%;酸杆菌门(Acidobacteria)、放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、绿菌门(Chlorobi)、蓝藻门(Cyanobacteria)、脱铁杆菌门(Deferribacter)、厚壁菌门(Firmicutes)、梭杆菌门(Fusobacteria)、芽孢单菌门(Gemmatimonades)、Gracilibacteria、硝化螺旋菌门(Nitrospirae)、Parcubacteria、浮霉菌门(Planctomycetes)、变形菌门(Proteobacteria)和柔膜菌门(Tenericutes)等15个优势门占细菌总量的97.09%以上。
15个优势菌门的菌群结构如图5所示,鱼体组中细菌在门水平上的菌群结构差异不大,占比前3的优势门为拟杆菌门、厚壁菌门和变形菌门,与饵料生物的门类细菌组成及其比例较为相似。经统计,健康幼鱼组共检测出8个门,占比前3的优势菌门比例依次为51.84%±0.63%、26.17%±0.84%和15.11%±0.40%,标准差均小于1%,表明健康幼鱼的菌群结构非常稳定;发病幼鱼组共检测到的细菌门数为19个,3种优势菌门的比例范围依次为22.31%~52.31%、18.77%~ 30.53%和14.93%~52.77%,组内比例波动较大。
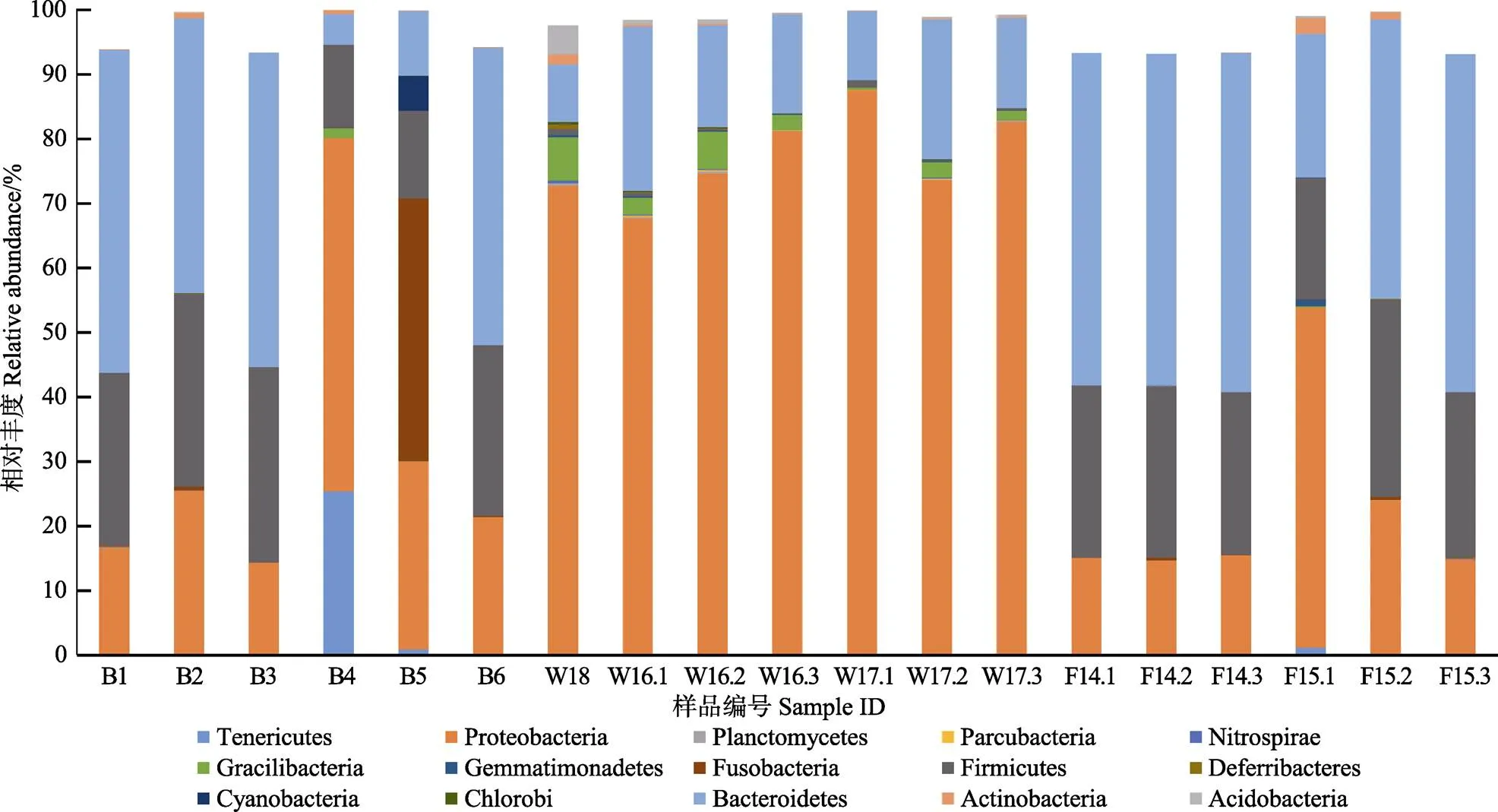
图5 各样品门水平下细菌相对丰度
基于OTU注释结果,丰度较高的15个优势菌科分别为弧菌科(Vibrionaceae)、Rhodobacteraceae、假交替单胞菌科(Pseudoalteromonadaceae)、紫单胞菌科(Porphyromonadaceae)、海洋螺菌科(Oceanospirillaceae)、支原体(Mycoplasmataceae)、毛螺菌科(Lachnospiraceae)、梭杆菌科(Fusobacteriaceae)、Flavobacteriaceae、科尔韦尔氏菌科(Colwelliaceae)、拟杆菌目某科(Bacteroidales_ S24_7_group)、拟杆菌科(Bacteroidaceae)、交替单胞菌科(Alteromonadaceae)、产碱杆菌科(Alcaligenaceae)和肠杆菌科(Enterobacteriaceae)。菌群结构如图6所示,15个科包含的细菌占总细菌含量的67.95%,样品间科水平的种类和细菌量各不相同。健康幼鱼组(Group_E)共检测出7个科,其中,至少在2个重复样品都检测到的科为6个,分别为紫单胞菌科(51.64%± 0.56%)、毛螺菌科(23.82%±1.03%)、产碱杆菌科(12.20%±0.32%)、肠杆菌科(2.89%±0.28%)、拟杆菌科(0.031%±0.019%)和拟杆菌目某科(0.022%±0.0097%),其6个科的细菌量占总细菌量的(90.59%±1.09%),与饵料组B1、B3和B6的菌群结构较为相似。与健康幼鱼组(Group_E)相比,发病幼鱼组(Group_F)组内菌属多样性和菌群结构差异均较大,F15.3菌群结构与健康幼鱼较为相似,而F15.1和F15.2中紫单胞菌科(2.71%±1.89%)、毛螺菌科(7.13%±5.12%)和产碱杆菌科(0.51%±0.10%)的细菌比重显著低于健康幼鱼组,肠杆菌科(18.81%± 2.41%)、拟杆菌科(11.78%±13.58%)和拟杆菌目某科(13.27%±4.29%)的细菌比重显著高于健康幼鱼组;除此之外,还有9个科只存在于发病幼鱼组,其中,至少在2个重复样品检测到的科有5个,分别为弧菌科(1.10%±0.11%)、Rhodobacteraceae (0.22%±0.30%)、支原体(0.48%±0.67%)、梭杆菌科(0.05%±0.03%)和Flavobacteriaceae (0.10%±0.13%)。
在属水平上,19个样品中细菌丰度最高的15个属分别为副杆菌属()、、、假交替单胞菌属()、大肠志贺氏杆菌属()、嗜冷菌属()、拟杆菌属()、科尔韦尔氏菌属()、、、弧菌属()、短波单胞菌属()、、冷杆菌属()、。
15种优势菌属在各样品中的菌群分布及结构特征见图7,健康幼鱼组组内属水平菌群结构稳定,含有5个菌属,分别为副杆菌属(59.13%±0.77%)、(25.03%±0.84%)、(13.97%±0.21%)、假交替单胞菌属(1.83%±0.18%)和拟杆菌属(0.036%±0.022%)。与健康幼鱼组相比,发病幼鱼组组内菌属多样性和菌群结构差异均较大,F15.3菌群结构与健康幼鱼较为相似,而F15.1和F15.2中的副杆菌属(2.57%±1.70%)、(0.88%± 0.42%)和(0.19%±0.12%)的细菌比重显著低于健康幼鱼组,拟杆菌属(13.27%±4.29%)和大肠志贺氏杆菌属(8.50%±2.11%)的细菌比重显著低于健康幼鱼组,除此之外,还有6个属只存在于发病幼鱼组,其中,至少在2个重复样品都检测到的属有4个,分别为弧菌属(0.31%±0.35%)、短波单胞菌属(4.25%±5.97%)、Olleya(0.03%±0.04%)和冷杆菌属(0.01%±0.01%)。

图6 各样品科水平下细菌相对丰度
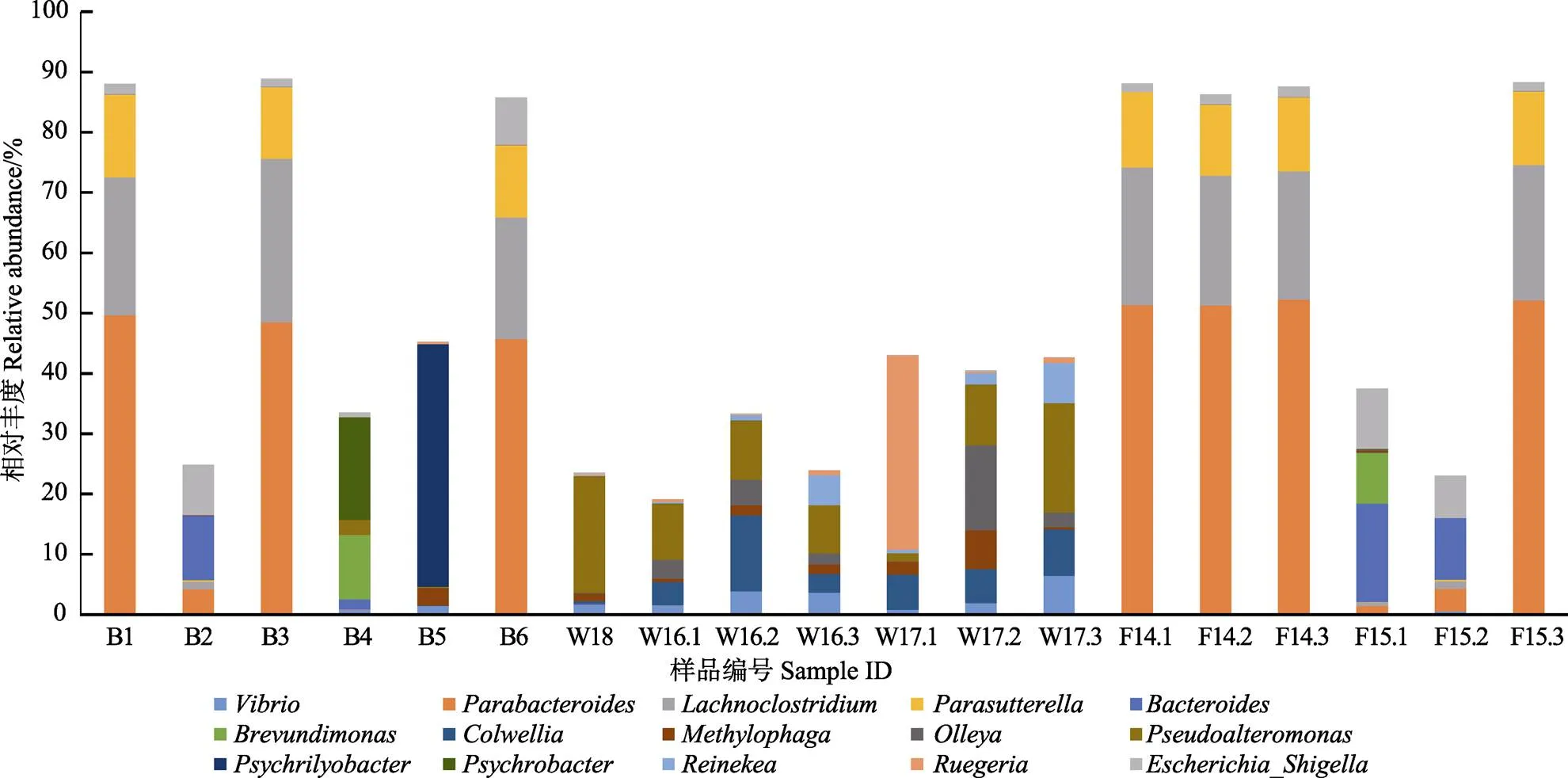
图7 各样品属水平下细菌相对丰度
2.3.4 丰度Top100的微生物的系统进化树分析
对OTU的丰度进行统计,选出丰度最高的100个OTU构建系统进化树,并以热图形式展示OTU在不同样品中的分布(图8)。经统计,这100条reads共归于 6个门: 变形菌门(63.54%)、拟杆菌门(21.88%)、厚壁菌门(8.33%)、梭杆菌门(1.56%)、蓝细菌门(1.56%)和Gracilibacteria(3.13%)。由图8可知,编号为F14.1、F14.2和F14.3的健康幼鱼和编号为F15.3的发病幼鱼与饵料组所属的金海湾小卤虫(B1)、西藏大卤虫(B3)和裂壶藻(B6)的高丰度OTU一致,主要为副杆菌属()、属、布劳特氏菌属()、属、梭菌目(Clostridiales)和奇异变形杆菌()等细菌。
编号为F15.1和F15.2的2条病鱼与生物饵料中无棣小卤虫(B2)、轮虫(B4)和小球藻(B5)中的高丰度OTU一致,主要为拟杆菌属()、拟杆菌目某科(Bacteroidales_S24_7_group)、肠球菌属()、约氏乳杆菌()、苍白杆菌属()、短波单胞菌属()、幽门螺杆菌()、肠杆菌属()、大肠志贺氏杆菌属()和奇异变形杆菌。
3 讨论
本研究利用Illumina Miseq平台对山东某育苗场暴发腹水症的30日龄大菱鲆幼苗及同期健康的幼苗、自然海水、养殖水体和生物饵料等共计19个样品内的细菌16S rDNA进行了高通量测序,所有样品OTU的Good’s coverages指数范围在0.93~1.00之间,说明测序深度足够大,数据可以全面解读各样品中细菌的组成及丰度,可以反映绝大多数的微生物信息。
对Simpson指数的计算可知,幼鱼、生物饵料和养殖水体的细菌丰度和多样性依次升高。有关半滑舌鳎()(张正等, 2015)和凡纳滨对虾()(孙振丽等, 2016)的研究结果与本研究结果一致,即养殖水体的微生物多样性比动物的肠道微生物多样性高。健康幼鱼和发病幼鱼共有的OTU分别占各自OTU总数的90.80%和10.17%。这说明健康的大菱鲆和发生腹水的大菱鲆肠道细菌多样性存在较大的差异,发病幼鱼会受到外界细菌的侵袭。为了溯源大菱鲆幼鱼肠道微生物,基于OTU对生物饵料、自然海水、健康幼鱼和发病幼鱼进行Venn图分析(图1),显示健康幼鱼和发病幼鱼分别与生物饵料共有的OTU个数为80和636,分别占幼鱼OTU总数的91.95%和81.85%。有研究认为,养殖环境中的微生物能够影响养殖动物肠道内的微生物多样性(Li, 2012),丰度较低的某些细菌会定植到鱼体肠道(Navarrete, 2009)。由此可知,水体的微生物多样性非常高,自然海水OUT的数目达到2124个,但其与健康幼鱼特有的OTU数为0,即使是与发病幼鱼特有的OTU数也仅为31。由此推测,在大菱鲆肠道微生物群落结构形成初期,生物饵料发挥了关键的作用。根据OTU注释结果,健康幼鱼的肠道微生物共聚类为8个门,发病幼鱼的肠道微生物可聚类为19个门。经统计,发病幼鱼组增加的11个门类细菌丰度均较小,细菌总量仅占3.91%±2.57%,如酸杆菌门、放线菌门、蓝藻门、Saccharibacteria门和螺旋藻门等。说明当幼鱼发病时,饵料中某些丰度较低的细菌会趁机植入肠道。

图8 基于丰度最高的100个OTU的系统进化树及其在各样品中的分布热图
B1: 金海湾小鲁虫; B2: 无棣小鲁虫; B3: 西藏大鲁虫; B4: 轮虫; B5: 小球藻; B6: 裂弧藻; W18: 自然海水; W16: 健康幼鱼养殖水体; W17: 发病幼鱼养殖水体; F14: 健康幼鱼; F15: 发病幼鱼
B1:in Jinhaiwan; B2:in Wudi; B3:in Tibet; B4:; B5:; B6:; W18: Sea water; W16: Aquaculture water of healthy juvenile fish; W17: Aquaculture water of diseased juvenile fish; F14: Healthy juvenile fish; F15: Diseased juvenile fish
根据细菌分类结果,拟杆菌门、厚壁菌门和变形菌门在大菱鲆幼鱼肠道中占优势地位。有学者研究了体长6 cm大菱鲆幼鱼(朱鹏飞, 2015)和体重700 g大菱鲆成鱼(邢孟欣等, 2014)的肠道菌群,显示变形菌门细菌是丰度最高的门类。而本研究对30日龄(体长为1 cm)健康幼鱼的肠道菌群研究结果显示,拟杆菌门是主要优势菌门,占细菌总量的50%以上;其中,变形菌门细菌仅占15.11%±0.40%。值得注意的是,与健康幼鱼相比,发病幼鱼中变形菌门的细菌丰度明显提高(30.56%±19.76%)。变形菌门因其细菌形态多样而得名,是细菌中最大的一个门,主要包括α-变形菌纲、β-变形菌纲、γ-变形菌纲、δ-变形菌纲和ε-变形菌纲,近年又确立一个ζ-变形菌纲(Emerson, 2007)。本研究发现,β-变形菌纲、伯克氏菌目、产碱杆菌科、属中的4种未知菌种广泛存在于健康大菱鲆幼苗体内,该属reads总数为12471条,占健康幼鱼体内变形菌纲细菌总量的80.79%。属细菌首次是从健康的人类粪便中分离出来(Nagai, 2009)。属的细菌是否参与大菱鲆幼苗正常的肠道代谢活动,还有待进一步研究。与健康幼鱼相比,发病幼鱼β-变形菌纲占比明显降低,仅占33.62%。
γ-变形菌纲是目前已知细菌种类最多的一个纲,包含许多能引起人类致病的细菌,如沙门氏菌属()能造成人类伤寒和肠炎。注释结果发现,健康幼鱼肠道含有的γ-变形菌纲的细菌较为单一,全部属于肠杆菌目肠杆菌科,占健康幼鱼变形菌纲细菌总数的19.12%;而发病幼鱼肠道中γ-变形菌纲的细菌较为多样,主要有肠杆菌目、交替单胞菌目(Alteromonadales)、Cellvibrionales目、着色菌目(Chromatiales)、海洋螺菌目(Oceanospirillales)、假单胞菌目(Pseudomonadales)、咸水球星菌目(Salinisphaerales)、硫发菌目(Thiotrichales)、弧菌目(Vibrionales)和黄色单胞菌目(Xanthomonadales)等,细菌丰度显著提高,占发病幼鱼变形菌纲细菌总数的51.19%。其中,肠杆菌科、假交替单胞菌目和弧菌目含有许多的致病菌。
对所有样品(饵料、水体和幼鱼)中丰度较高的100个OTU进行进化树–热图分析发现(图8),健康幼鱼肠道中高丰度菌属在饵料中的产自渤海的金海湾小卤虫、产自西藏的大卤虫和裂壶藻中中含量颇丰。这些菌属中大多属于环境或动物肠道正常菌群,除了少量奇异变形杆菌。奇异变形杆菌属于肠杆菌科,是一种条件致病菌,能引起大黄鱼()体表溃烂(张庆华等, 2005)、棘胸蛙()烂皮病(王瑞君等, 2012)和中华鳖()腮腺炎病(林亚歌等, 2104)等多种水产动物疾病。从图8也可看出,该菌在发病幼鱼生境内检测量有所提高。其次,发病幼鱼生境内高丰度菌属在饵料中的产自渤海的无棣小卤虫、轮虫和小球藻中颇丰。这些菌属中除了环境中广泛存在的化能自养类细菌和奇异变形杆菌外,还有一种致病菌属——大肠志贺氏杆菌属。大肠志贺氏杆菌属是一种肠道感染致病菌(刘蔚等, 1999),能引起人类痢疾(刘汉明, 1991)。而3种商品化卤虫和裂壶藻中大肠志贺氏杆菌属和奇异变形杆菌的含量都极其丰富。在生产上,卤虫需要摄食裂壶藻进行营养强化,然后才作为饵料投喂幼鱼,而裂壶藻和3种卤虫在门水平的菌群结构均较为相似,故推测鱼体内的大肠志贺氏杆菌属和奇异变形杆菌均来自裂壶藻。从细菌进化地位来说,大肠志贺氏杆菌属、奇异变形杆菌与迟缓爱德华氏菌同属于肠杆菌科(Enterobacteriaceae),而肠杆菌科除菊花欧文氏菌都含有肠杆菌科共同抗原(何晓青, 1980)。由此可知,发病大菱鲆幼鱼肠道除了增加许多的有害菌群外,其肠道含量较低的致病菌也存在丰度升高现象。
4 小结
本研究利用高通量测序技术对暴发腹水症的大菱鲆幼苗及其养殖环境的微生物多样性进行了研究。结果显示,生物饵料中的微生物参与大菱鲆幼鱼肠道早期菌群的形成,并起主要作用,也会引起发病幼鱼肠道有害菌群大量滋生。因此,为了有效预防大菱鲆幼鱼疾病的暴发,在育苗过程中,应规范养殖,减少应激,并加强开口饵料中病原菌的检查及防控力度。
Austin B, Zhang XH.: A significant pathogen of marine vertebrates and invertebrates. Letters in Applied Microbiology, 2006, 43(2): 119–124
Blanquer A, Alayse JP, Berrada-Rkhami O,. Allozyme variation in turbot () and brill () (Osteichthyes, Pleuronectoformes, Scophthalmidae) throughout their range in Europe. Journal of Fish Biology, 1992, 41(5): 725–736
Chen ZB, Li B, Wang ZB,. Study on the diversity of endophytic bacteria in Maize using Illumina MiSeq high- throughput sequencing system. Modern Food Science and Technology, 2016(2): 113–120 [陈泽斌, 李冰, 王定康, 等. 应用Illumina MiSeq高通量测序技术分析玉米内生细菌多样性. 现代食品科技, 2016(2): 113–120]
Dou Y, Zhao XW, Ding J,. Application of high-throughput sequencing for analyzing bacterial communities in earthen ponds of sea cucumber aquaculture in northern China. Oceanologia et Limnologia Sinica, 2016, 47(1): 122–129 [窦妍, 赵晓伟, 丁君, 等. 应用高通量测序技术分析北方刺参养殖池塘环境菌群结构. 海洋与湖沼, 2016, 47(1): 122–129]
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 2010, 26(19): 2460
Emerson D, Rentz JA, Lilburn TG,. A novel lineage of proteobacteria involved in formation of marine Fe-oxidizing microbial mat communities. PLoS One, 2007, 2(7): e667
He XQ. Sanitary inspection (bacterial examination) (second volume). Health and epidemic prevention station in Jiangxi Province, 1980 [何晓青. 卫生防疫检验(细菌检验)(下册). 江西省卫生防疫站, 1980]
Jing YY, Zhang Z, Wang YG,. Isolation and identification of pathogenic bacterium associate with ascites disease of cultured. Journal of Anhui Agricultural Sciences, 2016, 44(24): 106–108 [景亚运, 张正, 王印庚, 等. 养殖大菱鲆腹水病病原菌的分离与鉴定. 安徽农业科学, 2016, 44(24): 106–108]
Ligi T, Oopkaup K, Truu M,. Characterization of bacterial communities in soil and sediment of a created riverine wetland complex using high-throughput 16S rRNA amplicon sequencing. Ecological Engineering, 2014, 72: 56–66
Li HM, Bai L, Jiang DM,. Microbial diversity of piggery air detected by 16S rDNA high-throughput sequencing. Animal Science and Biotechology, 2015, 51(3): 81–84 [李红梅, 白林, 姜冬梅, 等. 基于16S rDNA高通量测序方法检测猪舍空气微生物多样性. 中国畜牧杂志, 2015, 51(3): 81–84]
Li J, Yan XH, Chen JX,. Studies on the characteristics of pathogenicisolated from diseased. Periodical of Ocean University of China (Natural Science), 2006, 36(4): 649–654 [李筠, 颜显辉, 陈吉祥, 等. 养殖大菱鲆腹水病病原的研究. 中国海洋大学学报(自然科学版), 2006, 36(4): 649–654]
Lin YG, Ye J, Shi TT,. Isolation and identification of pathogenfrom diseased Chinese soft-shelled turtle. Fisheries Science, 2014, 33(12): 800–803 [林亚歌, 叶键, 石婷婷, 等. 中华鳖源奇异变形杆菌的分离鉴定与致病性研究. 水产科学, 2014, 33(12): 800–803]
Li S, Sun L, Wu H,. The intestinal microbial diversity in mud crab () as determined by PCR-DGGE and clone library analysis. Journal of Applied Microbiology, 2012, 113(6): 1341–1351
Liu HM. Some advances in the epidemiology and molecular biology ofin China. Chinese Journal of Zoonoses, 1991, 7(2): 7–8 [刘汉明. 中国志贺氏菌属流行病学和分子生物学研究的某些进展. 中国人兽共患病学报, 1991, 7(2): 7–8]
Liu W, Zhao SH, Yang SQ,. Proper identification of. Practical Preventive Medicine, 1999, 6(1): 80–81 [刘蔚, 赵树红, 杨淑其, 等. 正确鉴定志贺氏菌属. 实用预防医学, 1999, 6(1): 80–81]
Nagai F, Morotomi M, Sakon H,.gen. nov., sp. nov., a member of the family Alcaligenaceae isolated from human faeces. International Journal of Systematic and Evolutionary Microbiology, 2009, 59(7): 1793
Navarrete P, Espejo RT, Romero J. Molecular analysis of microbiota along the digestive tract of juvenile Atlantic salmon (L.). Microbial Ecology, 2009, 57(3): 550–561
Pozhitkov AE, Beikler T, Flemmig T,. High-throughput methods for analysis of the human oral microbiome. Periodontology, 2011, 55(1): 70
Sun ZL, Xuan YM, Zhang H,. Bacterial diversity in theboone intestine and aquaculture environment. Journal of Fishery Sciences of China, 2016, 23(3): 594–605 [孙振丽, 宣引明, 张皓, 等. 南美白对虾养殖环境其肠道细菌多样性分析. 中国水产科学, 2016, 23(3): 594–605]
Thomson R, Macpherson HL, Riaza A,.biotype 1 as a cause of mortalities in hatchery-reared larval turbot,(L.). Journal of Applied Microbiology, 2005, 99(2): 243–250
Wang RJ, Xiong XJ. Isolation, identification and drug sensitivity tests offrom rotten-skin disease of. Freshwater Fisheries, 2012, 42(4): 31–34 [王瑞君, 熊筱娟. 棘胸蛙烂皮病奇异变形杆菌的分离、鉴定及对药物敏感性研究. 淡水渔业, 2012, 42(4): 31–34]
Wang XF, Zhao YF, Song XF,. Application of high- throughput sequencing techniques for analyzing bacterial communities in pond-raised mud crab () intestine and its aquaculture environment. Journal of Fishery Sciences of China, 2017, 24(6): 1245–1253 [王贤丰, 赵艳飞, 宋志飞, 等. 应用高通量测序技术分析拟穴青蟹肠道及其养殖环境菌群结构. 中国水产科学, 2017, 24(6): 1245–1253]
Xing MX, Li GY, Hou ZH,. Different taxonomic distribution of gastrointestinal tract microbiome of turbot (L.) individuals. Progress in Modern Biomedicine, 2014, 14(20): 3801–3805 [邢孟欣, 李贵阳, 侯战辉, 等. 不同大菱鲆()个体肠道菌群结构差异研究. 现代生物医学进展, 2014, 14(20): 3801–3805]
Zhang ML, Zhang MH, Zhang CH,. Pattern extraction of structural responses of gut microbiota to rotavirus infection via multivariate statistical analysis of clone library data. FEMS Microbiology Ecology, 2009, 70(2): 21
Zhang QH, Xiong QM, Xiao LL,. A pathogen isolated from skin-ulcer—ZXS02 strain. Journal of Fisheries of China, 2005, 29(6): 824–830 [张庆华, 熊清明, 肖琳琳, 等. 大黄鱼溃烂症的一种致病菌——奇异变形杆菌ZXS02菌株. 水产学报, 2005, 29(6): 824–830]
Zhang WN. Identification of a pathogen associated with swollen abdomen of cultured turbot and antigenicity of its outer membrane proteins. Master´s Thesis of Ocean University of China, 2006 [张伟妮. 大菱鲆腹水症病原菌的鉴定及其外膜蛋白的抗原性研究. 中国海洋大学硕士研究生学位论文, 2006]
Zhang XJ, Chen CZ, Fang H,.and its pathogen in the turbot (L.): A case report. Oceanologia et Limnologia Sinica, 2006, 37(5): 417–423 [张晓君, 陈翠珍, 房海, 等. 大菱鲆()病原鳗利斯顿氏菌的鉴定. 海洋与湖沼, 2006, 37(5): 417–423]
Zhang Z, Li B, Wang YG,. The microflora structure in digestive tract of half-smooth tongue sole (Günther) cultured in outdoor pond basing on high-through sequencing technique. Acta Hydrobiologica Sinica, 2015, 39(1): 38–45 [张正, 李彬, 王印庚, 等. 基于高通量测序的池塘养殖半滑舌鳎消化道菌群的结构特征分析. 水生生物学报, 2015, 39(1): 38–45]
Zhang ZD, Gu MY, Wang W,. Analysis of bacterial community in radiation polluted soils by high-throughput sequencing. Microbiology China, 2016
Zhu PF. Analysis of bacterial communities from turbot () aquaculture system and construction of DNA microarray detection system for aquaculture pathogenic bacteria. Master´s Thesis of First Institute of Oceanography, SOA, 2015 [朱鹏飞. 大菱鲆养殖环境细菌多样性分析及水产病原菌基因芯片检测体系的构建. 国家海洋局第一海洋研究所硕士研究生学位论文, 2015]
Turbot () Biodiversity Assessment Using High-Throughput Illumina Sequencing to Analyze Juvenile Turbot Intestines and Their Bacterial Cultures
WU Huanhuan1,2, WANG Weiji2, LÜ Ding2, HU Yulong2, KONG Jie1,2①
(1. Key Laboratory of Exploration and Utilization of Aquatic Genetic Resources, Ministry of Education; College of Fisheries and Life Science, Shanghai Ocean University, Shanghai 201306; 2. Key Laboratory of Sustainable Development of Marine Fisheries, Ministry of Agriculture and Rural Affairs, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao 266071)
In order to study the effects of environmental factors on the intestinal flora structure of turbot (), we used high-throughput sequencing to explore the bacterial community structure and diversity in juvenile turbot intestines, the culture environment, and biological baits. The results showed that 547621 effective sequences were detected in nineteen samples, and they could be classified into 3771 operational taxonomic units (OTUs), among which 3038, 1090, 87, and 777 originated from the aquaculture water, the biological baits, healthy juvenile turbot intestine, and diseased juvenile fish intestine, respectively. There were 57 OTUs shared between the healthy juvenile turbot intestine and the biological baits, 0 OTU shared between the healthy juvenile turbot intestine and aquaculture water, 481 OTUs shared between the diseased juvenile fish intestine and the biological baits, 31 OTUs shared between the diseased juvenile fish intestine and the aquaculture water. The effect of biological bait on microbial diversity of intestinal tract of juvenile fish was much greater than that of environment. In total, the predominant phyla in the turbot intestine were Bacteroidetes, Firmicutes, and Proteobacteria. The intestinal microflora of healthy juvenile turbot can be clustered into 8 phyla, and the intestinal microflora of the diseased juvenile fish could be clustered into 19 phyla. Compared with the healthy juveniles, the community structure of the predominant phyla was imbalanced at the intestinal level of the diseased juvenile fish. Furthermore, analysis of the 100 most abundant bacterial OTUs in the different samples revealed that the species dominant in the intestinal bacteria of juvenile fish was closely related to the dominant species in the biological baits. Meanwhile, the intestinal dominant bacteria species of each diseased juvenile are different. This study provided the basis for healthy culture and micro-ecological regulation of turbot.
Biodiversity; High throughput sequencing; Turbot ()
KONG Jie, E-mail: kongjie@ysfri.ac.cn
S968
A
2095-9869(2019)04-0084-11
10.19663/j.issn2095-9869.20180507003
* 山东省农业良种工程项目“泰山学者种业计划专家项目”(ZR2014CQ001)和山东省农业良种工程项目“大菱鲆种质资源精准鉴定与选种育种创新利用-子课题”(2016LZGC031-2)共同资助 [This work was supported by Taishan Scholar Program for Seed Industry (ZR2014CQ001), and Accurate Identification and Selection Breeding Creative Utilization of Turbot Germplasm Resources (2016LZGC031-2)]. 吴欢欢,E-mail: 17664080328@163.com
孔 杰,研究员,E-mail: kongjie@ysfri.ac.cn
2018-05-07,
2018-07-09
吴欢欢, 王伟继, 吕丁, 胡玉龙, 孔杰. 应用高通量测序技术分析大菱鲆幼鱼肠道及其养殖环境的微生物群落结构. 渔业科学进展, 2019, 40(4): 84–94
Wu HH, Wang WJ, Lü D, Hu YL, Kong J. Turbot () biodiversity assessment using high-throughput Illumina sequencing to analyze juvenile turbot intestines and their bacterial cultures. Progress in Fishery Sciences, 2019, 40(4): 84–94
(编辑 冯小花)
