一个常染色体显性遗传非综合征型聋家系MYO6基因新致病性变异△
2019-07-23田涛鲁雅洁姚俊曹新魏钦俊李琦
田涛 鲁雅洁 姚俊 曹新 魏钦俊 李琦,3
据估计,约70%的遗传性听力损失(hereditary hearing loss, HHL)是非综合征型(nonsyndromic autosomal hearing loss, NSHL),其余30%被归类为综合征型听力损失[1,2]。常染色体隐性遗传类型的NSHL(autosomal recessive NSHL,ARNSHL)通常表现为语前聋,约占听力损失患者的80%,而常染色体显性遗传NSHL(autosomal dominant NSHL,ADNSHL)大部分是渐进性语后听力损失,约占20%[3]。目前至少有100个耳聋相关基因已被鉴定,其中包括36个ADNSHL相关基因(http://hereditaryhearingloss.org,09/2017)。由于ADNSHL多为散发且家系罕见,其HHL的分子诊断仍然具有挑战性[4]。
以往耳聋家系致病基因的鉴定主要采用候选基因筛选和连锁分析定位克隆等技术,复杂而且时间较长,存在一定的局限性。目前,全外显子组测序已成为阐释HHL复杂性的最合适和最可靠的工具[5~7]。故本研究采用全外显子组测序调查一个中国ADNSHL家系(JSNY-067)的遗传病因,发现了MYO基因的新致病性变异,报告如下。
1 资料与方法
1.1家系资料 一个6代共52人的ADNSHL大家系(图1,编号为JSNY-067家系),家系调查和听力学检测由南京医科大学附属儿童医院耳鼻咽喉科完成。本研究经南京医科大学人类研究所伦理委员会批准,所有家系成员已经书面签字同意赫尔辛基宣言的所有原则。
该家系六代居住于江苏省,14例表现为语后ADNSHL,均为双侧对称的感音神经性听力损失,听阈图表现为平坦型或者高频下降型;听力损失程度似乎与个体的年龄相关,家系中听力损失者均主诉听力障碍在学龄时出现,随着年龄增加听力损失呈进行性加重,早期为中高频听力损失,逐渐影响所有频率呈平坦型听阈曲线,两名年龄最小的患者(5岁和10岁患者)感音神经性听力损失仅影响中低频,其中2例(Ⅳ3、Ⅴ3)典型听阈图见图2。
所有听力损失者其他耳鼻咽喉科专科检查均正常,无耳毒性药物应用史和噪声暴露史,无主观和客观的前庭功能障碍表现,无耳鸣,鼓室导抗图正常,全身检查神经系统正常,未发现视力障碍和视网膜病变。
1.2DNA提取和全外显子组测序 抽取所有参与调查家系成员的外周静脉血,按照标准程序提取基因组DNA。使用DNA提取试剂盒(DP319,Tiangen,Beijing,China)从受试者的血液中提取基因组DNA,使用耳聋基因变异检测试剂盒(Capital Bio Corporation,Beijing,China )检测中国人群四种常见耳聋基因(GJB2、SLC26A4、GJB3和MT-RNR1)中的9种热点致病性变异,包括:GJB2的35delG、176del16、235delC、299delAT, GJB3的538C>T,SLC26A4的2168A>G、919-2A>G, MT-RNR1的1494C>T、1555A>G[8]。

图1 JSNY-067家系的家系图 黑色箭头是先证者

图2 2例典型听力损失患者(Ⅳ3、Ⅴ3)的听阈图 平均听阈(pure tone audiometry,PTA)分别为58.3 dB HL(Ⅳ3)、41.7 dB HL(Ⅴ3)
参照说明书, 将约3 μg纯化的gDNA片段化至200 bp,对文库制备进行末端修复,腺苷酸化和衔接子连接。使用TruSeq DNA LT / HT样品制备试剂盒和TruSeq Exome Enrichment Kit富集外显子组,合并等量的文库样品,然后与定制的捕获阵列杂交,包括外显子、剪接位点和立即融合内含子序列。 在Illumina HiSeq 2500上进行测序,用于变异鉴定和随后的分析。 Samtools mpileup识别变体并标记SNP和插入缺失,ANNOVAR用于注释基因。
1.3Sanger测序和变异功能分析 运用Sanger测序对JSNY-067家系的所有成员的样品进行测序以确定变异基因中的致病性变异,尤其是MYO6基因致病性变异。 使用BigDye Terminator v3.1循环测序试剂盒(Applied Biosystems,Foster City,CA,USA)对PCR产物进行测序,并使用ABI 3700XL测序仪进行测序分析。MYO6测序的引物如下:MYO6-正向:5'TGTCCCGCCCATGTTAATAT 3'; MYO6反向:5'TGCACCGTACCTCAACACAG 3'。
1.4变异位点保守性分析 使用SIFT、Polyphen2、PROVEAN和MutationTaster软件来确定影响表型的蛋白质结构的可能变化。使用Clustal X1.83软件比较人类野生型MYO6蛋白序列与来自黑猩猩、小家鼠、抹香鲸、野猪、食蟹猴、双峰骆驼、鼠耳蝙蝠、埃及果蝠、牛、大熊猫、白豚、八齿鼠、原鸡和热带爪蟾的MYO蛋白序列(序列见http://www.ensembl.org/),并研究这种蛋白质的进化保护和结构预测。
2 结果
2.1JSNY-067家系听力损失特点 JSNY-067家系中14例语后ADNSHL患者中1例(Ⅲ10)已经去世,未测听力,其余13例的听力损失发生年龄及特点见表1,可见,听力损失程度随年龄增长呈逐渐加重的趋势,年龄越小,听力损失程度越轻,年龄越大,听力损失程度越重。
2.2基因检测结果 为了搜寻耳聋候选基因,本研究对该家系的一例听力正常(Ⅲ13)个体和两例听力损失(Ⅳ3和Ⅴ3)个体的DNA进行了全外显子组测序。 每例个体平均生成46.7亿个碱基,目标区域的平均测序深度约为97个,以满足单核苷酸多态性(SNPs)和插入缺失的要求。 将测序数据与NCBI人参考基因组进行比对,并与包含来自1000基因组计划试验数据的dbSNP138,来自八个测序的HapMap个体和来自YH数据库的10个个体进行比较。
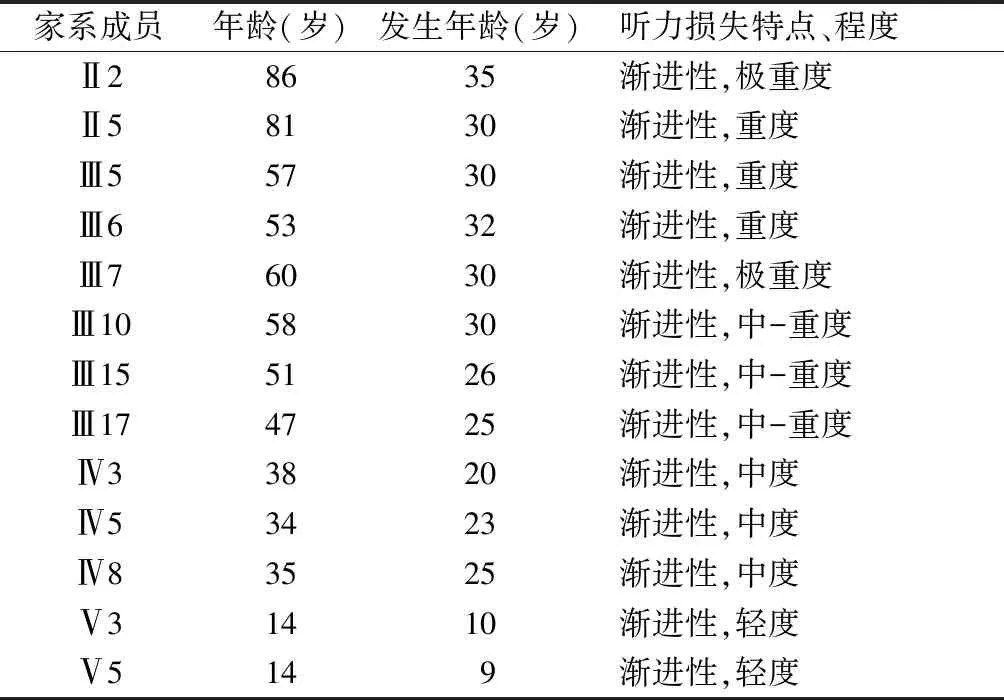
表1 JSNY-067家系13例听力损失患者的听力损失发生年龄、特点及程度
在2例个体总共发现了7 898种变异,其中5 760个是非同义变异,包括剪切位点变异、点变异以及插入缺失。 然后将这些变异根据以下原则进行筛选:①变体的等位基因频率cuto(在dbSNP138,HapMap,1000基因组和本地数据集中小于0.01);②在所有受影响的个体中发现的变异,在未受影响的个体中没有发现,进一步将候选变异缩减为25个错义变异。
通过Sanger测序筛选谱系样本中发现的25个变异,在MYO6基因的外显子8中发现了与该疾病共分离的错义变异c.622A> G(p.K208E)(图3),该变异在该家系中13个听力损失成员中发现,但在21个听力正常的家系成员中未发现。 为了评估其是否为致病性变异,进一步测序了500个汉族健康人和300个散发性耳聋病例,均未发现c.622A> G变异。
2.3变异位点保守性分析结果 为了预测该变异的可能致病性,本研究分析了氨基酸替换的进化保守性和破坏性效应, 结果显示,该变异在多种脊椎动物物种间高度保守,MYO6蛋白中208位的Pro残基在多种脊椎动物物种中高度保守(图3b),当这个位置改变时,引起致病性变异的可能性大大增加。进一步在计算机中使用SIFT,Polyphen2,PROVEAN和MutationTaster软件预测了变异危害的可能性,预测分析的结果见表2。MYO6基因编码非常规肌球蛋白VI,可以分为三个区域:N-末端运动区域,接头结构域和单个IQ基序组成的颈部结构域,以及具有卷曲螺旋结构的C末端尾区域。p.K208E变异可能影响驱动子部位的截短蛋白(图3b),表明,MYO6新的变异c.622A> G(p.K208E)可能是这个家系发生听力损失的原因。

表2 JSNY-067家系MYO6基因的致病性变异特性
注:a.得分范围从0(有害)到1(中性),截止分数设置为0.05。b.负分和正分分别表示有害和中性,截止分数设置为-2.5。 评分等于或低于-2.5的变异被认为是“有害的”,高于-2.5被认为是“中性的”。c.HGMD(Human Gene Mutation Database,人类基因变异数据库)

图3 JSNY-067家系MYO6基因的变异分析 a.野生型和c.622A> G变异对照的DNA序列。b.由运动、IQ和卷曲螺旋结构域组成的肌球蛋白VI的图结构, 显示p.K208E变异在驱动子头部区域中映射,保守性分析表明,肌球蛋白VI蛋白中208位的Pro残基在多种脊椎动物物种中保守
3 讨论
听力损失的基因诊断目前已经广泛应用,但是由于遗传异质性使得分子诊断昂贵而耗时。全外显子组测序是鉴定孟德尔遗传中的致病基因变异的有效方法,与Sanger测序相比,具有明显的成本效益性[9]。本研究对一个中国耳聋家系进行研究,该家系听力损失患者表现为渐进性的语后聋,符合ADNSHL的遗传学特征,通过对该家系进行全外显子组测序,成功鉴定了导致该家系迟发性听力损失的新的MYO6变异c.622A> G(p.K208E)。
MYO6基因位于染色体6q14.1,含有35个外显子,构成3 858个多核苷酸编码区[10]。已知MYO6基因中的变异可以引起ADNSHL(DFNA22)和ARNSHL(DFNA37)。 迄今为止,全世界许多国家的不同DFNA22家族报道了11种MYO6致病变异[11],MYO6致病 变异似乎在外显子2~35中自由分布,只有两个突变(p.Arg205Stop,p.Arg205Gln)位于p.K208E附近,临床表型均不是ADNSHL。MYO6基因编码非常规肌球蛋白-VI,其是一种反向运动蛋白,向肌动蛋白丝的负端移动,并在细胞内囊泡和细胞器运输中发挥作用[12]。 蛋白质由含有ATP-和肌动蛋白结合位点的运动区域和与其他蛋白质相互作用的球状尾巴组成[13]。 肌球蛋白-VI有助于维持内耳毛细胞的结构完整性和适当的功能,MYO6错义变异可能抑制野生型肌球蛋白VI二聚化并抑制其功能[14]。
在本研究JSNY-067家系中,先证者(Ⅴ3)具有双侧对称的感音神经性听力损失,听阈图呈高频下降型,10岁左右开始发病,2例年龄最小的受影响受试者(5岁和10岁)感音神经性听力损失仅影响中低频,随后听力损失渐进性发展,受影响的家庭成员表现为早期仅高频听力下降,随着年龄增加全频听力下降。染色体6q13上的MYO6基因的外显子8中鉴定发生了错义变异c.622A> G,这导致用蛋白质残基208 (K208E)处的谷氨酸置换赖氨酸。在500例汉族健康人和300例散发耳聋病例均未发现p.K208E变异,考虑MYO6基因的c.622A>G(p.K208E)变异是JSNY-067家系的致病变异,该变异型肌球蛋白Ⅵ可能导致ADNSHL,其影响蛋白质的致病性还有待进一步研究。
