(4-咔唑-9-基-苯亚甲基)-(5-咔唑-9-亚甲基-[1,3,4]噻二唑-2-基)-胺的合成与表征
2019-07-19王金玉
王金玉
(中国人民解放军61699 部队,湖北 枝江 443200)
咔唑因含有的共轭体系较大和分子内强的电子转移,具有的给电子能力和空穴传输能力较强,及热稳定性和光化学稳定性较高[1-3];其衍生物在光电材料[4]、染料[5]、医药[6]、超分子识别[7]、化学传感[8]等领域具有潜在的广泛应用。因此,合成含咔唑基结构化合物对新药及新材料的研究和开发都具有十分重要的意义。
更为重要的是咔唑类化合物易进行功能团的结构修饰,其3 位,6 位和9 位易引入,导致咔唑衍生品具有许多独特的性能及生物活性,且在紫外光范围有很强的吸收,并带隙在3.2eV 左右及发蓝光等特性[3-11],其原料容易得到。同时,研究表明具有大范围共轭结构的电子給体能有效提高光敏材料的发光特性。
因此,为了探讨新型的大范围共轭结构的咔唑类化合物,本文以咔唑、氯乙酸、对氯苯甲醛、硫代氨基脲为原料,经反应合成5-咔唑-9-亚甲基-2-氨基-1,3,4-噻二唑和4-咔唑-9-基苯甲醛,然后在多聚磷酸催化剂作用下经亲核加成-消除反应得到了(4-咔唑-9-基苯甲醛)-(5-咔唑-9-亚甲基-[1,3,4]噻二唑-2-基)-胺,其结经IR,1H NMR 表征。其合成路线如图1。
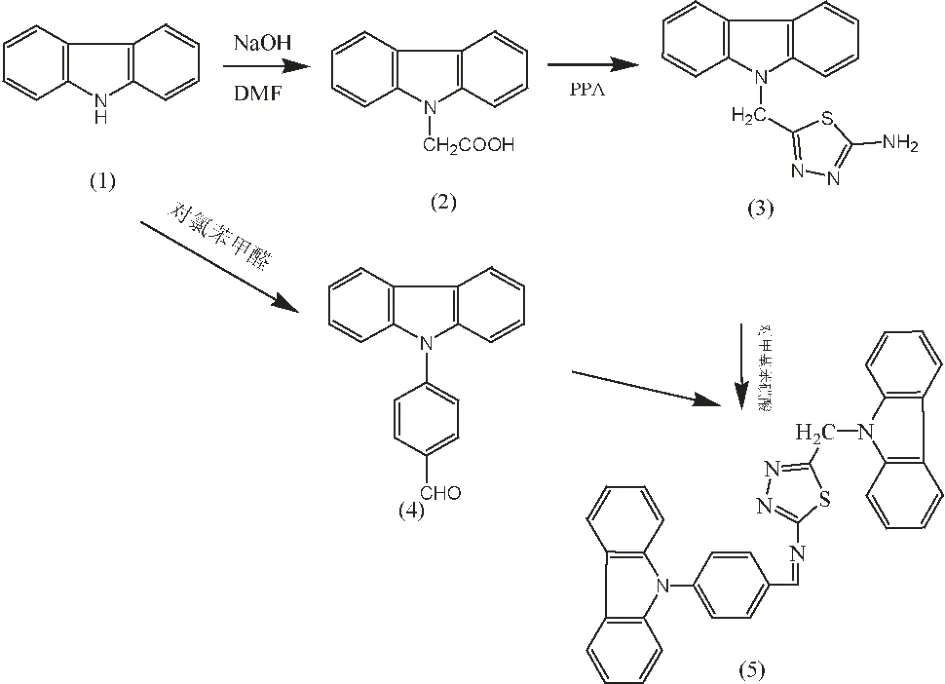
图1 (4-咔唑-9-基-苯亚甲基)-(5-咔唑-9-亚甲基-[1,3,4]噻二唑-2-基)-胺的合成路线
1 实验部分
1.1 仪器与试剂
X-4 型显微熔点测试仪;UV-2100 型紫外可见光光度计(1cm 石英比色皿,纯溶剂标定基线);Bruke AM-80 型核磁共振仪(CDCl3为溶剂,TMS 为内标);FT-IR 型红外光谱仪(KBr 压片);Perkin-Elmer240 型元素分析仪。
试剂:咔唑(化学纯,天津市光复精细化工研究所);氯乙酸(分析纯,天津市福晨化学试剂厂);硫代氨基脲(分析纯,天津市科密欧化学试剂有限公司);对氯苯甲醛(分析纯,北京市百灵威有限公司);其余所用试剂均为分析纯或化学纯。
1.2 合成或制备
1.2.1 N-咔唑乙酸(2)的合成
在100mL 三颈瓶中,按投料比1∶1.2∶3 依次加入咔唑(2g,12mmol),氢氧化钠(1.44g,36mmol),其后加入30mLDMF,升温至80℃并保持此温度下回流反应,至氢氧化钠溶解完,再缓慢加入氯乙酸(1.36g,14.4mmol)。由TLC 检测至原料咔唑反应完全(乙酸乙酯∶石油醚=1∶5)。反应结束后,倒入冰水中并不断搅拌至有大量白色固体生成,减压过滤,滤液用浓盐酸调pH 至1-2,并不断搅拌,有大量的白色固体生成,减压过滤,滤饼自然烘干得粗产品。将粗产品用无水乙醇进行重结晶,干燥后得到N-咔唑乙酸,白色粉末固体2.64g,即得到目标产物,产率为98%。m.p.185~188℃。(文献值[12]184~184.5℃)
IR(KBr 压片) ν/cm-1:3417,3048,2814,1668,1597,1491,1451,1388,1236,994,857,750,721.
1.2.2 5-咔唑-9-亚甲基-2-氨基-1,3,4-噻二唑(3)的合成
空间调制系统的ML检测算法中,hlx需要4Nr次实数乘法,计算矢量的范数需要2Nr次实数乘法,由于ML检测要搜索所有的天线和符号,因此ML检测的计算复杂度为CML= 6MNtNr[13].
在100mL 三颈瓶中,按投料比1∶1 依次加入N-咔唑乙酸(0.56g,2.5mmol),硫代氨基脲(0.23g,2.5mmol),20mL 的PPA,升温至80℃并保持此温度下回流反应,由TLC 检测(二氯甲烷∶甲醇=1∶10),冷却至室温,在冰水浴下加入适量的水,再次升温至120℃,并保持此温度下回流反应8h。反应结束后,在冰水浴下用氨水调pH至9-10,有大量的固体生成,减压过滤,滤饼烘干,得到粗产品。将粗产品用95%的乙醇进行重结晶,得到白色粉末固体0.52g,即目标产物,产率为75.3%。m.p.121~123℃。(文献值[13]M.p119~120℃)
IR(KBr 压片) ν/cm-1:3274,3185,3045,2813,2723,1598,1456,1347,1220,1001,849,740.
1.2.3 4-咔唑-9-基苯甲醛(4)的合成
在带有搅拌棒和回流冷凝管的100mL 三颈瓶中,按 投 料 比1 ∶1 ∶1.2 依 次 加 入 咔 唑(0.5g,3mmol),对氯苯甲醛(0.38g,3mmol),三乙胺(0.36g,3.6mmol),溶于15mL 的DMF 中,同时开启加热器和搅拌器,加热至80℃并保持此温度下反应20h,由TLC 检测(乙酸乙酯∶石油醚=1∶5)。反应液冷却至室温,倒入冰水中,产生大量的固体,减压抽滤,滤饼自然烘干,得到粗产品。将粗产品用无水乙醇和丙酮的混合液进行重结晶,得到黄色粉末固体0.71g,产率为88%。m.p.138~140℃。(文献值[14]m.p.134~136℃)
IR(KBr 压片) ν/cm-1:3050,2817,2727,1698,1598,1491,1447,924,850,751.
1.2.4 目标化合物(5)的合成
向干燥的研钵中按投料比例为1∶1∶1.2 依次加入0.0002mol 的4-咔唑-9-基-苯甲醛0.05g,0.0002mol 的5-咔唑-9-亚甲基-2-氨基-1,3,4-噻二唑0.05g 和0.00024mol 的对甲基苯磺酸0.03g,室温下研磨20min 至均匀,TLC 检测(乙酸乙酯∶石油醚=1∶3),得到混合物。将混合物置于烘箱中,在75~80℃的温度下保温1h,待混合物冷却后,用体积浓度为90%的乙醇洗涤、抽滤,得到的滤饼,将滤饼用无水乙醇重结晶,即得目标产物。白色固体0.08g,产率为86%。m.p.239~240℃。
1H NMR(400 MHz,CDCl3) δ 8.11(s,1H) ,7.55(s,1H),7.47(s,1H),7.14 (d,J=8.7 Hz,2H),6.99(d,J=8.5 Hz,1H),6.95(s,2H),6.91(d,J=7.0 Hz,4H) ,6.89-6.88 (m,1H) ,6.77(s,1H),6.75(s,1H),6.7(s,8H),6.56 (d,J=8.4 Hz,2H),4.97(s,2H).
Anal.calcd for C34H24N5S:C 74.71,H 4.30,N 12.45,O 2.84;found C 74.57,H 4.21,N 12.14,O 2.68。
2 结果与讨论
2.1 催化剂用量对产品收率的影响
以5-咔唑-9-亚甲基-2-氨基-1,3,4-噻二唑和4-咔唑-9-基苯甲醛为原料,研究了5-咔唑-9-亚甲基-2-氨基-1,3,4-噻二唑与对甲基苯磺酸的物质的量比对反应产率的影响,分别选了1∶0.5、1:0.8、1∶1、1∶1.2、1∶1.5 进行了实验,其数据见表1。
由表1 中看出,随着投料的物质的量比的逐渐减小,反应产率逐渐增加并趋于稳定。这是由于在目标化合物的合成是满足pH=3~5 的条件下,在投料的物质的量比大于1∶1 的时候,对甲基苯磺酸的用量较少,导致反应体系的酸性较弱,5-咔唑-9-亚甲基-2-氨基-1,3,4-噻二唑中的氨基的质子化因碱性而减弱,反应速率减慢;当投料的物质的量比达到1∶1 的时候,研磨大概10min,反应体系有水产生。这是由于随着对甲基苯磺酸的用量增加,反应体系的酸性逐渐达到pH=3~5,5-咔唑-9-亚甲基-2-氨基-1,3,4-噻二唑中的氨基全部成为游离的氨基,完成了脱去氢离子的催化反应。继续减小投料的物质的量比,反应产率的增加趋于平衡。综合考虑,5-咔唑-9-亚甲基-2-氨基-1,3,4-噻二唑与对甲基苯磺酸的物质的量比选用1∶1~1.2 之间。

表1 反应物与对甲基苯磺酸的物质的量比对反应产率的影响Table 1 Effect of mole ratio on the yield
2.2 反应时间对反应收率的影响
最后研究了反应时间对产率的影响,在无溶剂的条件下,于干燥研钵中加入等摩尔的5-咔唑-9-亚甲基-2-氨基-1,3,4-噻二唑以及4-咔唑-9-基苯甲醛,以对甲基苯磺酸作为催化剂,室温下快速研磨,尝试不同的研磨时间,研究了不同的研磨时间对产率的影响,其结果见表2。

表2 反应时间对产率的影响Table 2 Effect of reaction time on the yield
从表2 看出,时间太短,原料反应不完全,目标化合物的产率很低,而随研磨时间的延长,配体的产率急剧增加。当研磨20min 时,反应最充分,所得产率也最高。而再继续延长反应时间,随着副产物的增加,配体产率有所下降。因此,最合适的研磨时间为20min。
2.3 谱图表征
(4-咔唑-9-基苯甲醛)-(5-咔唑-9-亚甲基-[1,3,4]噻二唑-2-基)-胺含有咔唑环本身的共轭,咔唑环与苯环的共轭及-C=N 键与取代环的共轭效应。为了考察化合物结构光谱的影响,我们测定了目标化合物在DMF 溶液中的紫外-可见吸收光谱,如图1 所示。

图1 (4-咔唑-9-基苯甲醛)-(5-咔唑-9-亚甲基-[1,3,4]噻二唑-2-基)-胺的UV-Vis 谱图Fig.1 The UV-Vis spectrum of(4-carbazole-9-ylbenzomethylene)-(5-carbazole-9-methylene-[1,3,4]thiadiazole-2-yl)-amine
如图1 所示,目标化合物在DMF 溶液中UV吸收光谱分别在300 和340nm 附近有明显的吸收带,最大紫外-可见吸收峰波长为295nm,其330nm 和345nm 处有两处吸收峰稍微较弱。从化合物分子结构看出,因咔唑分子骨架本身的π-π共轭结构在295nm 处出现最大的吸收峰,在330nm 和345nm 波长处的吸收带是因为苯环和咔唑结构的π-π 共轭及噻二唑与-C=N 结构的p-π共轭而产生的。由此可以看出合成的产物有很好的光物理性质。

图2 (4-咔唑-9-基苯甲醛)-(5-咔唑-9-亚甲基-[1,3,4]噻二唑-2-基)-胺的红外光谱图Fig.2 The FT-IR spectrum of (4-carbazole-9-ylbenzomethylene)- (5-carbazole-9-methylene-[1,3,4]thiadiazole-2-yl)-amine
由图2 知,3077cm-1左右处出现的较弱的特征峰为苯环C-H 的伸缩振动吸收峰;饱和C-H即咔唑环上的亚甲基的特征吸收峰出现在2924cm-1、2851cm-1处;在1602cm-1处产生很强的吸收振动峰,说明醛上C=O 与5-咔唑-9-亚甲基-2-氨基-1,3,4-噻二唑上的NH2进行缩合反应,产生C=N 基团;苯环骨架振动吸收峰的位置 出 现 在1512cm-1,1453cm-1,1381cm-1;在1031cm-1左右处的特征峰为C-C 的伸缩振动吸收峰;芳环外C-H 面外弯曲振动吸收峰出现在924cm-1处;C-S-C 的特征吸收峰出现在747cm-1 左右处;出现在747cm-1,649cm-1的特征峰为苯环取代产生的振动伸缩吸收峰。以上说明5-咔唑-9-亚甲基-2-氨基-1,3,4-噻二唑与4-咔唑-9-基苯甲醛缩合反应成功,得到(4-咔唑-9-基苯甲醛)-(5-咔唑-9-亚甲基-[1,3,4] 噻二唑-2-基)-胺。
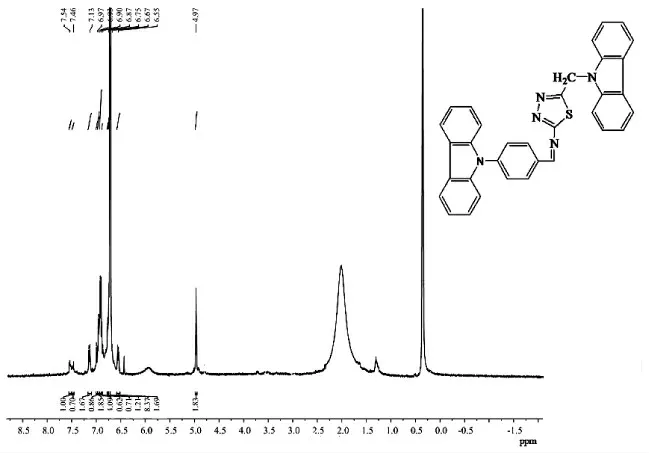
图3 (4-咔唑-9-基苯甲醛)-(5-咔唑-9-亚甲基-[1,3,4]噻二唑-2-基)-胺的1H NMR 图Fig.3 The 1H NMR spectrum of (4-carbazole-9-ylbenzomethylene)- (5-carbazole-9-methylene-[1,3,4]thiadiazole-2-yl)-amine
由图3 看出,出现在4.97ppm 处为希夫碱的咔唑基团连接的-CH2-上的信号位置;芳环上H的化学信号出现在6.55~7.56ppm 之间;受-C=N-键的影响,出现在8.11ppm 之间处的吸收峰为与-C=N-键相连的H 的化学信号;由此证明合成了目标化合物(4-咔唑-9-基苯甲醛)-(5-咔唑-9-亚甲基-[1,3,4]噻二唑-2-基)-胺。
3 结论
本文以咔唑、氯乙酸、硫代氨基脲和对氯苯甲醛为初始原料,通过液相回流法得到5-咔唑-9-亚甲基-2-氨基-1,3,4-噻二唑和4-咔唑-9-基苯甲醛,这两种化合物通过无溶剂研磨法合成了大共轭新的含咔唑基噻二唑类化合物,通过红外光谱、1H NMR 进行了结构表征。在DMF 溶液中测量了化合物的UV-vis,发现具有很好的光物理性质。同时研究了反应条件,得出结论:反应物物质的量比为1∶1∶1.2,反应时间为20min 时,反应产物的产率达到86%。
