盐酸d3-Poziotinib 的设计合成及体外肝微粒体稳定性研究
2019-07-19邵明莎马术超彭祥福白信法张少云
邵明莎,马术超,彭祥福,白信法,张少云,姚 雷,
(1.烟台大学新型制剂与生物技术药物研究山东省高校协同创新中心、分子药理和药物评价教育部重点实验室(烟台大学),山东 烟台 264005;2.绿叶制药集团有限公司,山东 烟台 264003)
Poziotinib (图1,HM781-36B)是一种pan-HER 酪氨酸激酶抑制剂,目前处于II/III期临床研究阶段[1-5],可单独或联合使用,用于胃癌[6]、非小型细胞性肺癌[7]及乳腺癌[8]的治疗.临床研究中发现,Poziotinib在体内的主要代谢途径是通过肝药酶代谢生成活性代谢产物M2(图1).而M2被认为是该药产生主要毒副反应如严重腹泻,呕吐等的主要原因.
封闭代谢位点是一种常见的改善药物代谢稳定性的修饰策略.通过封闭药物的代谢位点,可以延缓药物的清除速率,阻断活性代谢物的生成,延长药物在体内的作用时间.

图1 Poziotinib和M2的结构
氘是氢的稳定性非放射性同位素,氘的重要特点是其在药物分子中的形状和体积与氢基本上相同.因此,氘代药物一般会保留原来药物的生物活性和选择性.但由于碳-氘键的强度大于碳-氢键,氘代药物在药理学方面有以下几个优点:(1)降低体内清除率,增加药物的生物半衰期,从而减少副作用增强疗效;(2)降低药物在胃肠道或肝脏的代谢,调高药物的耐受性;(3)延长药物在体内的作用时间,增强药物疗效.目前,首个FDA批准上市的氘代药物是Concert 制药公司的用于治疗囊性纤维化的孤儿药 CTP-656.该药是氘代化的Ivacaftor,氘化后,由于半衰期的延长,给药次数变成1天给药1次,简化了治疗方案[9-10].2017年,Teva公司的氘代丁苯那嗪(Austedo)被批准用于治疗亨廷顿舞蹈症相关的舞蹈病症状[11].
基于以上事实, 在保持Poziotinib药理活性的前提下,为减少或封闭不良代谢,提高药物的耐受性,本课题组设计合成了盐酸d3-Poziotinib.
1 实验部分
1.1 试剂与仪器
3,4-二氯-2-氟苯胺(玛雅试剂);CD3I(Aldrich),N-Boc-哌啶-4-醇(百灵威科技有限公司),7N氨的甲醇溶液(Aldrich),丙烯酰氯(安耐吉化学), NADPH+(Acros),SGW-X4型显微熔点仪(上海精密科学仪器有限公司);Bruker ACF-400型核磁共振波谱仪(瑞士Bruker公司);FTIR Nicolet Impact 410型红外光谱仪(美国Nicolet); HPLC (Agilent 1200);expressionsCMS (美国Advion公司);Agilent G6530A.
1.2 化合物的合成路线
以已知化合物4-氯-7-羟基-6-特戊酰氧基喹唑啉(1,图2)为原料,在碱性条件下,与氘代碘甲烷反应生成中间体2.中间体2中的氯原子首先被2-氟-3,4-二氯苯胺取代,然后在氨的甲醇溶液作用下,脱掉特戊酰保护基得到中间体3.中间体3在碱性条件下与N-Boc-4-哌啶-对甲苯磺酸酯(TSP,4)发生醚化反应得到中间体5.中间体5在浓盐酸作用下,脱掉Boc保护基团后,直接与丙烯酰氯反应,得到丙烯酰胺衍生物,随后该衍生物再与盐酸成盐即可得到盐酸d3-Poziotinib(6)[14-15].

图2 d3-Poziotinib的合成路线(1)
另外,课题组后期按照图3所示路线也得到了目标化合物,即氘代化合物3′在碱性条件下,可与已知化合物7发生分子间亲核取代反应,生成d3-Poziotinib,然后与浓盐酸成盐可制备盐酸d3-Poziotinib,此方法也是可行的,较路线(1)简捷,但路线(1)所用原料经济易得,较适合工业化大规模生产,所以选择路线(1)进行展开实验.
1.3 合成步骤
(1)N-Boc-哌啶-4-对甲苯磺酸酯(TSP,4)按文献[12]方法合成.白色固体,mp:97.90 ~ 98.70 ℃.


图3 d3-Poziotinib的合成路线(2)
(3)4-氯-7-羟基喹唑啉-6-基特戊酸酯(1)按文献[13]方法合成.白色固体.
(4)d3-4-氯-7-甲氧基喹唑啉-6-基特戊酸酯(2)的合成:将化合物1(4.50 g, 16.10 mmol)与K2CO3(6.70 g, 48.30 mmol)溶于DMF(50.00 mL)中,控制温度在0 ℃下,滴加CD3I(1.50 mL, 24.10 mmol).加毕,反应体系在室温下搅拌16 h,至TLC检测反应显示原料消耗完毕.往体系中加入少量水,乙酸乙酯萃取(3×100 mL),合并有机相,无水硫酸钠干燥,过滤,有机相浓缩,残余物用硅胶柱层析纯化,得到2.90 g白色粉末状固体,收率60.60 %.1H NMR (CDCl3, 400 MHz)δ: 8.95 (s, 1H, ArH), 7.86 (s, 1H, ArH), 7.40 (s, 1H, ArH), 1.42 [s, 9H, (CH3)3CO].
(5)d3-4-(3,4-二氯-2-氟苯胺)-7-甲氧基喹唑啉-6-基特戊酸酯(3)的合成:将化合物2(2.90 g,9.76 mmol)与3,4-二氯-2-氟苯胺(2.10 g, 11.70 mmol)溶于乙腈(20 mL)中,加热90 ℃,搅拌反应8 h.TLC检测反应完毕后,将反应体系冷却至室温.抽滤,固体用乙腈(2×20 mL)洗涤,常温干燥过夜,得产品为白色固体2.89 g,收率67.40%.滤液浓缩后经硅胶柱纯化,得回收部分产品.mp:267.10~267.30 ℃.1H NMR (CDCl3, 400 MHz)δ: 11.16 (brs, 1H, ArNH), 8.78 (s, 1H, ArH), 8.43 (s, 1H, ArH), 7.67 (s, 1H, ArH), 7.50 ~ 7.54 (t, 1H,J=8.2 Hz, ArH), 7.20 ~ 7.23 (dd, 1H,J=8.7, 1.5 Hz, ArH), 1.52 [s, 9H, (CH3)3CO].
(6)1-叔丁氧羰基-4-(4-(3,4-二氯-2-氟苯胺基)-7-甲氧喹唑啉-6-基氧基)哌啶(5)的合成:将化合物3(2.89 g, 6.60 mmol)溶于少量甲醇中,冰浴条件下,滴加7 N氨的甲醇溶液(30 mL).滴加完毕,在室温下搅拌反应体系10 h,至TLC检测反应完全后.抽滤,固体用DCM(2×10 mL)洗涤,常温干燥,得产品0.60 g为黄色粉末状固体,收率为98.90 %.未经纯化可直接用于下一步.
将上述化合物(0.60 g, 1.70 mmol)、TSP(4)(1.21 g, 3.40 mmol)与K2CO3(0.70 g, 5.10 mmol)溶于DMF(4 mL)中,加热至67 ~ 70 ℃,反应24 h至TLC 检测原料消耗完毕.体系冷却至室温,缓慢滴加水(4.80 mL),室温反应3 h,抽滤,固体用水(2×10 mL)洗涤,常温干燥,得产品5为类白色至淡黄色粉末状固体8.00 g,收率43.60 %.1H NMR (d6-DMSO, 400 MHz)δ: 9.6 (brs, 1H, ArNH), 8.39 (s, 1H, ArH), 7.86 (s, 1H, ArH), 7.58 (m, 2H, ArH), 7.23 (s, 1H, ArH), 4.68 ~ 4.73 (m, 1H, OCH), 3.67 ~ 3.71 (m, 2H, NCH2), 3.25 (m, 2H, NCH2), 1.98 ~ 2.01 (m, 2H, CH2), 1.57 ~ 1.67 (m, 2H, CH2), 1.42 [s, 9H, OC(CH3)3].
(7)盐酸d3-Poziotinib (6)的合成:将化合物5(8.00 g, 14.90 mmol)溶于丙酮(80 mL)中,向其中滴加浓盐酸(14.40 mL),室温反应5 h,TLC检测反应完毕后,抽滤,固体用丙酮(2×10 mL)洗涤,常温干燥,得中间体为黄色固体7.40 g,收率97.60 %.未经纯化可直接用于下一步.将上述黄色固体(8.70 g, 17.00 mmol)溶于THF(61 mL)中,在剧烈搅拌下,室温滴加NaHCO3(5.71 g, 68 mmol)的水溶液87 mL.控温在0 ~ 3 ℃下,滴加丙烯酰氯(1.60 mL, 19.90 mmol)的THF(61 mL)溶液.滴加完毕,反应体系在该温度下搅拌反应30 min至TLC检测反应完毕.向反应体系中加入水140 mL,以乙酸乙酯(3×100 mL)萃取,合并有机相,有机相以无水硫酸钠干燥,过滤,浓缩.残余物经硅胶柱纯化,得d3-Poziotinib的游离碱为淡黄色泡沫状固体5.93 g,收率70.70 %.直接用于下一步.将上述d3-Poziotinib的游离碱(5.80 g, 11.70 mmol)在加热条件下,溶于甲醇(50 mL)中.稍冷,往体系中滴加浓盐酸至体系呈酸性(pH值2 ~ 3),室温搅拌24 h,抽滤,收集固体,固体用丙酮(3×10 mL)洗3次.得盐酸d3-Poziotinib为淡黄色至黄色粉末状固体4.60 g.收率:74.20 %. mp:230~232 ℃(分解).1H NMR (d6-DMSO, 400 MHz)δ: 12.0 (brs, 1H, ArNH), 8.83 (s, 1H, ArH), 8.57 (m, 1H, ArH), 7.59 ~ 7.67 (m, 2H, ArH), 7.36 (s,1H, ArH), 6.81 ~ 6.87 (m, 1H, ArH), 6.81~ 6.13 (dd,J=16.68, 2.32 Hz,1H, ArH), 5.67 ~ 5.70 (dd,J=10.44, 2.32 Hz,1H, ArH), 5.02 (m, 1H, OCH), 3.87 ~ 3.99 (m, 2H, NCH2), 3.55 ~ 3.67 (m, 2H, NCH2), 2.08 ~ 2.11(m, 2H, CH2), 1.57 ~ 1.67 (m, 2H, CH2).13C NMR (DMSO-d6,100 MHz) 164.3, 158.9, 157.3, 154.8, 152.3, 149.0, 147.8, 135.8, 131.2, 128.4, 127.8, 127.2, 125.7(d), 125.2(d), 120.2(d), 107.0, 106.1, 100.2, 73.7, 42.5, 40.1, 30.9, 30.0. ESI-MS,m/z:494.0 [M+H]+.
1.4 盐酸d3-poziotinib 体外肝微粒体稳定性
取肝微粒体溶液2.00 mL (浓度为1.00 mg/mL),加入试验药物溶液2.00 μL (浓度5μmol/L),混匀,每样品平行3份,至37 ℃水浴中孵育3 min,加入10 μL 400 mmol/L NADPH再生液(1.30 mmol/L NADP+, 3.3 mmol/L葡萄糖-6-磷酸, 3.30 mmol/L氯化镁, 0.40 U/mL葡萄糖-6-磷酸),开始计时,温孵0 min, 5 min, 15 min, 30 min, 1 h,2 h时,取样100 μL至200 μL的冰甲醇终止反应,放入-20 ℃冰箱待测.
HPLC色谱条件:色谱柱:Capcellpak C18 UG120-4.6*150 mm 5 Micron;流动相A:12.50 mmol/L KH2PO4+50 mmol/L NaClO4H2O pH值 2.5; 流动相B:100%乙腈; A∶B=41∶59 (V/V).
2 结果与讨论
2.1 化合物的结构
盐酸Poziotinib与d3-Poziotinib 在结构上仅有一个甲基的区别,因此,在核磁中,2种化合物除有无甲基信号峰外,其他峰信号应均一致.图4 对比了盐酸Poziotinib (a) 与d3-Poziotinib(b)的1H NMR和13C NMR.1HNMR图谱中,Poziotinib在化学位移为4 附近有一个甲基的单峰信号,而d3-Poziotinib在此位置无信号;同样,在13C NMR图谱中,Poziotinib在化学位移为56.59 附近有一个甲基信号峰,而d3-Poziotinib在此位置无信号.质谱检测显示d3-Poziotinib的 MS(ESI):494.0 [M+H]+,411.0,357.0;HRMS: C23H18D3Cl2FN4O3: 493.116 3, 实测493.117 1[M+H]+,进一步验证了结构的正确性.
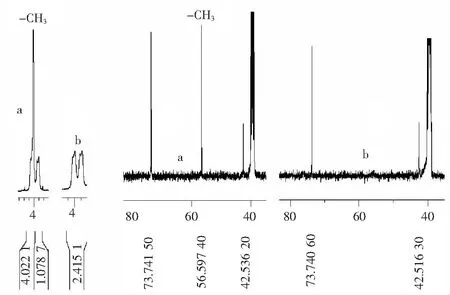
图4 盐酸Poziotinib(a)与d3-Poziotinib (b)的核磁对比
2.2 化合物的体外肝药酶稳定性实验
通过内标法,检测在上述时间点测试药物的剩余量,结果见图5.从表中可以看出在1 h 和2 h时,Poziotinib的剩余量分别是84 % 和72 %,而d3-Poziotinib 的剩余量分别是89 %和79 %.同时通过推算,Poziotinib和d3-Poziotinib 在大鼠中的半衰期分别为3.5 h和4.6 h.从此可以看出与原药Poziotinib相比,d3-Poziotinib 具有一定的代谢稳定性及较长的半衰期.

图5 Poziotinib与d3-Poziotinib的药物剩余量比较
Fig.5 The drug residue comparison of Poziotinib andd3-Po-ziotinib
3 结 论
本研究以已知化合物4-氯-7-羟基-6-特戊酰氧基喹唑啉为起始原料,经7步反应合成盐酸d3-Poziotinib,总收率为9.02 % (条件未经优化).该合成路线操作简单,条件温和,适于工业化生产.同时,体外的肝药酶稳定性实验显示盐酸d3-Poziotinib的半衰期较Poziotinib有一定延长,这也为进一步研究其体内药代动力学行为及药效学奠定了良好的基础.
