Biomarkers and subtypes of deranged lipid mettabolism in non-alcohlic fatty liver disease
2019-07-10JosMatoCristinaAlonsoMazenNoureddinShellyLu
José M Mato, Cristina Alonso, Mazen Noureddin, Shelly C Lu
Abstract Nonalcoholic fatty liver disease (NAFLD) is a heterogeneous and complex disease that is imprecisely diagnosed by liver biopsy. NAFLD covers a spectrum that ranges from simple steatosis, nonalcoholic steatohepatitis (NASH) with varying degrees of fibrosis, to cirrhosis, which is a major risk factor for hepatocellular carcinoma. Lifestyle and eating habit changes during the last century have made NAFLD the most common liver disease linked to obesity,type 2 diabetes mellitus and dyslipidemia, with a global prevalence of 25%.NAFLD arises when the uptake of fatty acids (FA) and triglycerides (TG) from circulation and de novo lipogenesis saturate the rate of FA β-oxidation and verylow density lipoprotein (VLDL)-TG export. Deranged lipid metabolism is also associated with NAFLD progression from steatosis to NASH, and therefore,alterations in liver and serum lipidomic signatures are good indicators of the disease's development and progression. This review focuses on the importance of the classification of NAFLD patients into different subtypes, corresponding to the main alteration(s) in the major pathways that regulate FA homeostasis leading, in each case, to the initiation and progression of NASH. This concept also supports the targeted intervention as a key approach to maximize therapeutic efficacy and opens the door to the development of precise NASH treatments.
Key words: S-adenosylmethionine; Methionine adenosyltransferase; Lipid metabolism;Multiomics; Lipidomics; Nonalcoholic steatohepatitis; One-carbon metabolism; Very low-density lipoproteins; Steatosis; Precision medicine
INTRODUCTION
Fat storing is common to many different species. The desert locust stores lipids in the“fat body”, a dynamic tissue that plays an essential role in energy storage and utilization in insects[1], to migrate from south-western Morocco to the Iberian Peninsula covering a distance of 600 miles without settling down. Some fish also store fat for their survival. Without eating and powered only by stored fat, salmons swim 2000 miles up the fresh waters of the Yukon River from the Bering Sea to reach their spawning grounds. Long distance migrating birds, such as the bar-tailed godwit, the ruby-throated hummingbirds and the bar-headed geese, accumulate large amounts of fat prior to departing. Likewise, the gray whale increases its fat stores prior to swimming more than 10000 miles between feeding grounds in the Artic to the nursery lagoons of Mexico´s Baja Peninsula; and hibernating mammals such as the grizzly bears, after a period of incomparable hyperphagia, do not eat for 5 to 7 mo subsisting solely on stored fat.
The energy source for these prodigious feats are fatty acids (FA) stored as triglycerides (TG) into lipid droplets (LD) primarily in the adipose tissue and liver.The mobilization of FA from adipose tissue TG stores requires the activity of TG lipases that generate FA, which are then released into the blood and taken up by hepatocytes, where are reincorporated into TG (Figure 1). Some of these re-esterified TG combine with apolipoprotein-B (APOB) to form very low-density lipoproteins(VLDL), and are exported into circulation. This process is regulated by microsomal TG transfer protein (MTTP) and accompanied by encapsulating the neutral lipid core with a phospholipid (PL) monolayer enriched in phosphatidylcholine (PC) molecules containing polyunsaturated FA (PUFA), such as arachidonic acid (AA; 20:4n-6) and docosahexaenoic acid (DHA; 22:6n-3)[2,3]. APOB, cholesterol and other apolipoproteins(like APOC) are also found decorating the surface of the VLDL-TG particle[2,3]. The largest amount of TG used for the synthesis of VLDL (VLDL-TG) is synthesized from FA entering the liver from the adipose tissue, even under conditions where the synthesis of FA from glucose and fructose by de novo lipogenesis (DNL) is high (see below). Humans preferentially oxidize carbohydrate over fat, a process that helps to maintain blood glucose homeostasis. Most of the TG in circulation during the postabsorptive phase are associated with VLDL-TG[2]. This mechanism uncouples hepatic TG synthesis (energy storing) from TG secretion and maintains a low blood content of FA, which are cytotoxic.
NONALCOHOLIC FATTY LIVER DISEASE
TG are energy dense and chemical stable compounds. By weight, FA provide more than twice as much energy (9 kcal/g) as carbohydrates and proteins (4 kcal/g), and match the caloric density of diesel (8 kcal/g). From this perspective, fatty liver may be considered a physiological adaptation and an evolutionary advantage to anticipate periods of prolonged food (energy) shortage. However, lifestyle and eating habit changes during the last century have made fatty liver the most common liver disease linked to obesity, type 2 diabetes mellitus (T2D) and dyslipidemia, with a prevalence of 25%[4-7]. Nonalcoholic fatty liver disease (NAFLD) covers a spectrum that ranges from simple steatosis (NAFL), nonalcoholic steatohepatitis (NASH) with varying degrees of fibrosis, to cirrhosis, which is a major risk factor for hepatocellular carcinoma (HCC). NASH is distinguished from steatosis by the presence of inflammation and hepatocyte injury. Approximately 25% of individuals with NAFL progress to NASH. Of those that develop NASH, 25% progress to cirrhosis, of whom at least 1%-2% per year develop HCC[4-6]. NASH is now the leading cause of liver transplantation in women[8]and projected to be the leading indication in the United States by 2020[4-6]. Degree of liver fibrosis is the major factor linked to all-cause mortality[9]. However, NAFLD does not always follow an orderly progression. For instance, it is possible for NAFLD patients to develop fibrosis without going through the NASH stage, or to develop liver cancer despite absence of fibrosis or histologic NASH[4-6,10]. Studies have reported 10-70% of HCC cases in NAFLD occurred without cirrhosis[11]. The annual direct medical cost is > $100 billion in the United States alone for NAFLD[4-6]. Despite the huge investment by the pharmaceutical industry there are still no approved therapies targeting NASH[12]. Lifestyle changes are the only therapeutic strategy that can halt the progression of NAFLD[4-6]. Clearly, both a better understanding of the factors that promote progression from simple steatosis to NASH, fibrosis and liver cancer is sorely needed to improve our therapeutic strategy.

Figure 1 Lipid metabolism. The mobilization of fatty acids (FA) from their triglyceride (TG) storage in the adipose tissue is promoted by TG lipases. The resultant FA are then released into the blood and taken up by hepatocytes.Other sources of hepatic FA are the dietary lipids in chylomicrons and de novo lipogenesis induced by carbohydrates.These FA are metabolized by mitochondrial or peroxisomal β-oxidation, accumulated in the cytoplasm inducing lipotoxicity, or subsequently elongated, desaturated and re-esterified for synthesis of complex lipids such us phospholipids (PL), diglycerides or TG. Some of the re-esterified TG are packed into very low-density lipoproteins combined with apolipoprotein-B and exported into circulation. This process is regulated by microsomal triglyceride transfer protein and accompanied by encapsulating the neutral lipid core with a PL monolayer enriched in phosphatidylcholine molecules containing polyunsaturated FA. Enzyme reactions regulated by S-adenosylmethionine(SAMe) and pathways in which SAMe deficiency may lead to the accumulation of TG and progression to nonalcoholic steatohepatitis are indicated in blue. APOB: Apolipoprotein-B; DG: Diglycerides; ER: Endoplasmic reticulum; FA:Fatty acids; MTTP: Microsomal triglycerides transfer protein; PC-PUFA: Phosphatidylcholines containing polyunsaturated fatty acids; PL: Phospholipids; SAMe: S-adenosylmethionine; TG: Triglycerides; VLDL: Very lowdensity lipoproteins.
LIVER LIPID METABOLISM IN NAFLD
Consisting with its energy storage function, the relationship between the intrahepatic TG (IHTG) content and VLDL-TG secretion rate is curvilinear. In subjects with normal IHTG (up to 5% of liver weight), VLDL-TG export increases linearly with IHTG content; but in individuals with steatosis, VLDL-TG secretion reaches a plateau independently of the amount of IHTG[13,14]. Genetic defects (APOB, APOC3, MTTP,TM6SF2) that impair hepatic VLDL-TG secretion cause hepatic steatosis that may progress to NASH with fibrosis, even without obesity or T2D[15-19]; and impaired APOB synthesis has been observed in NASH patients as compared to obese controls[20]. These results indicate that a reduction in the capacity to export VLDL-TG increases the risk to develop NASH. Consistently, patients treated with antisense APOB or MTTP inhibitors, which lower VLDL assembly and secretion, are associated with hepatic steatosis, inflammation and fibrosis, which limit their utility[21,22]. The discovery that the effect of defective VLDL-TG secretion extends well beyond the management of liver energy storage to promote the development of NASH and fibrogenesis emphasizes the importance of identifying therapeutic targets for NASH reversal in the setting of impaired VLDL-TG secretion. It is important to note,however, that the increase in susceptibility to develop NASH in obese subjects that are carriers of the TM6SF2 E167K variant, which impairs VLDL-TG export, is accompanied by protection from cardiovascular disease due to the reduced serum levels of atherogenic lipoproteins[23]. This is important when designing treatments that aim to increase VLDL-TG export in NASH.
Hepatic steatosis arises when the uptake of FA and TG from circulation and DNL saturate the rate of FA β-oxidation (in the mitochondria and peroxisomes) and VLDLTG export (Figure 1). NAFLD subjects often show an increase in DNL[13,24], and it has been proposed by many that DNL is a major pathway in the pathogenesis of NAFLD[25]. On this premise the pharmacological inhibition of DNL that include (1)Downregulating SREBP-1c, the major transcriptional regulator of the enzymes involved in DNL, (2) Decreasing the activity of the DNL rate-limiting enzyme,specifically acetyl-CoA carboxylase (ACC), and (3) Inhibiting stearoyl-CoA dehydrogenase 1 (SCD1), the first irreversible step committing FA to TG synthesis, are being studied in phase 2 and 3 clinical trials of NASH[26]. However, a potential limitation of this approach is that a decrease in DNL may induce an increase in FA uptake to the liver from circulation, the major source of hepatic lipids, or a decrease in FA oxidation as compensatory mechanisms[27,28]. From an evolutionary stand point, it seems unlikely that an increase in DNL would be a major pathway in the development of NAFLD. FA from the adipose tissue and from the diet contribute about 59% of TG in the livers of patients with NAFLD, while DNL contributes 26% of intrahepatic FA, and dietary TG transported by chylomicrons 15% of liver fat[29].Accordingly, the inhibition of liver FA uptake has been shown to improve NASH in experimental models[30]; albeit at the risk of increasing FA in circulation, peripheral FA stores, and weight gain, which may limit its potential therapeutic application. The importance of increased DNL in NASH development should, however, not be minimized since increased DNL may just as well overwhelm a deficient VLDL-TG exporting system which, presumably, is already saturated caused by increased hepatocellular lipid uptake. The increase in DNL in NAFLD may be an adaptive mechanism for the generation of metabolic signals that direct lipids toward beneficial pathways to improve energy balance even in the setting of excess FA accumulation, a concept known as lipoexpediency (the antonym to lipotoxicity[31,32]). For instance, it has been shown that FA synthase, the DNL enzyme that catalyzes the conversion of acetyl-CoA to the 16-carbon FA palmitate, is involved in the activation of PPARα (an activator of FA oxidation that is expressed at high concentrations in the liver) via the synthesis of its ligand, palmitoyl-stearoyl-phosphatidylcholine (PC-16:0/18:1)[33].NAFLD subjects also show an increase in the rate of hepatic FA oxidation[34,35]because of mitochondrial uncoupling between FA oxidation and ATP synthesis[36]. Increased FA oxidation in NAFLD may be, however, detrimental to the liver due to the excessive generation of reactive oxygen species. Together, these results suggest that different individuals (NAFLD subtypes) could have different alterations in the major pathway(s) that regulate FA homeostasis leading to NAFLD[37]. Evidence from clinical trials indicating that only a small percent (20%-50%) of NASH patients benefit from the different treatments supports this concept[26]. Thus, the identification of noninvasive metabolic biomarkers that would allow the classification of patients into different subtypes that correspond to the main alteration(s) leading to the initiation and progression of NASH would be of great help for the development of precise treatments.
S-ADENOSLYMETHIONINE AS A LINK BETWEEN LIPID METABOLISM AND HEPATOCELLULAR ONE CARBON METABOLISM
Assessing the hepatic lipid metabolism, it is important to note that LD are not only critically important for energy metabolism in terms of TG storage, but are also a major supply of (1) PL precursors, such as diacylglycerols (DG) and other lipids of the monoalk(en)yl diacylglycerol family, that give rise to diacyl-PL and plasmalogens,respectively; (2) Cholesterol, which is stored as cholesteryl-esters (CE); and (3) FA, not only saturated FA, such as palmitate (16:0) and stearate (18.0), which are cytotoxic,but also PUFA, such as AA, that gives rise to the eicosanoid family of inflammatory mediators (prostaglandins, thromboxanes, and leukotrienes), and DHA, which is antiinflammatory[38]. The main lipid classes found in the core of liver LD are TG, DG, and CE, which are enveloped by a PL monolayer (mainly made of PC) decorated with proteins that are important in lipid remodeling, signaling and energy storing[39,40]. PC found in LD are synthesized both by the Kennedy route, whose last step is the reaction of CDP-choline with DG to form PC and cytidine monophosphate; and the PE N-methyltransferase (PEMT) pathway, which converts PE rich in PUFA (mainly AA and DHA) into PC through three successive N-methylations of the PE amino group, with S-adenosylmethionine (SAMe) as the methyl donor[41](Figure 1). SAMe is a versatile molecule which is the source of essentially all methyl transfer reactions in cells[42]. Liver plays a central role in SAMe metabolism, as this is where up to half of the daily intake of methionine is catabolized via its conversion to SAMe[43]. This reaction is catalyzed by methionine adenosyltransferase (MAT). Two genes encode for MAT, MAT1A is expressed in normal differentiated liver and MAT2A is expressed in all extrahepatic tissues as well as in fetal liver[43]. In liver, SAMe homeostasis is controlled by MAT-mediated synthesis and utilization, largely accomplished by glycine N-methyltransferase (GNMT)[43](Figure 2). Accordingly, GNMT deletion in mice induces a massive increase in intrahepatic SAMe content[44]that accelerates the flux of methyl groups through multiple pathways, including PEMT and DNAmethylation, leading to aberrant liver lipid signatures, development of NASH, fibrosis and HCC[45].
SAMe metabolism is coupled to the folate cycle and together they form the so called one carbon metabolism (1CM) (Figure 3). 1CM circulates 1-carbon units from different nutritional and amino acids inputs (choline, betaine, folate, glucose,methionine, serine, glycine and threonine), via SAMe and 5-methyltetrahydrofolate(MTHF), into a large variety of outputs, such as PL-, protein- and DNA-methylation,and glutathione (GSH), polyamines, reduced nicotinamide adenine dinucleotide phosphate (NADPH), and nucleotide synthesis, that regulate key biological processes ranging from VLDL-TG export, gene expression and redox homeostasis, to DNA synthesis and cell growth. Mat1a knockout (KO) mice have chronically low hepatic SAMe level (75% lower)[46], show reduced content of PC-PUFA (mainly AA and DHA)[37]and, as expected, impaired synthesis and release of VLDL-TG, which leads to the accumulation of TG, DG and FA, accumulation of oxidized FA, oxidative stress,and abnormal hepatic lipid signatures, which trigger the spontaneous development of steatosis and its progression to NASH, fibrosis and HCC[37,43,46]. In Mat1a KO mice, low SAMe also associates with increased serum levels of amino acids methionine, serine and glycine; increased hepatic MTHF, decreased GSH content, and altered protein and DNA methylation[37]. MAT1A is often downregulated in NAFLD patients with more advanced fibrosis[47]. Consistently, several studies showed human NASH have reduced transmethylation[48], hepatic PC/PE ratio[49], and abnormal VLDL-TG assembly and export[50]. These results suggest that SAMe deficiency may be a critical driver of NASH in a subgroup of NAFLD patients. Importantly, SAMe treatment of the Mat1a KO mice after onset of NASH for two months corrected many of the abnormalities, nearly normalized the liver histology, and reduced blood ALT, AST and TG levels without altering cholesterol content[37]. SAMe treatment of rats fed a methionine and choline deficient (MCD) diet, which reduces hepatic SAMe content and induces steatohepatitis, also improved liver histology[51]. Taken together, these results support the concept that 1) a reduction in SAMe is a common driver of NAFLD initiation and progression to NASH in humans, and 2) that NAFLD patients with M-subtype serum metabolomic profile (see below) will likely benefit from SAMe treatment, but this has not yet been examined.
CIRCULATING BIOMARKERS OF NAFLD
The advent of lipidomics has taught us that each lipid class (e.g., TG, PC) is made of a multitude of different lipid molecular species varying in the length and number of double bonds of their FA chains[52,53]; and that the lipid homeostatic status is implemented by a large family of FA desaturases and elongases in conjunction with lipases, acyl-transferases, PL and sphingolipid synthesizing enzymes, and phospholipases[54,55]. Changes in lipid signatures (lipid molecular species compositions) can have profound effects on cell function, regulating processes such as oxidative phosphorylation[56]. A sequence variant in PNPLA3 that is strongly associated with NAFLD has been related to TG remodeling and VLDL-TG secretion in hepatocytes[57,58], suggesting that abnormal lipid remodeling may be key to the development and progression of NAFLD. Accordingly, mice modify the liver lipid profile in response to a variety of conditions that induce steatosis and its progression to NASH, such as ablation of methionine adenosyltransferase 1A (Mat1a)[46], fasting or feeding a high fat diet[41], or feeding an MCD diet[59]. It has also been observed that the serum lipidomic profile reflects the liver lipidome[37], a finding which supports the search of noninvasive NAFLD biomarkers in blood. At present, liver biopsy is the“gold standard” to diagnose NASH, an invasive, imprecise and expensive procedure with possible complications. As a result, numerous studies have been published aiming to the identification of panels of circulating biomarkers (using genomics,transcriptomics, proteomics and metabolomics) for steatosis, NASH and fibrosis diagnosis, as well as for risk prediction of NAFLD progression and response to therapy[60,61]. Some studies have shown that lipidomic patterns can differentiate between normal liver and NAFLD[62,63]. Interestingly, recent studies also focus on the discrimination between simple steatosis and NASH[64]or the detection of advance fibrosis[65]. However, a burning challenge in NAFLD research is the identification of which patients with NAFLD will develop NASH and, for those with NASH, how fast the disease will progress. At present, it is premature to conclude which of these blood biomarkers, alone or in combination, would be best to precisely and rapidly diagnose the severity of NASH and monitor the liver's response to treatment[60,61].
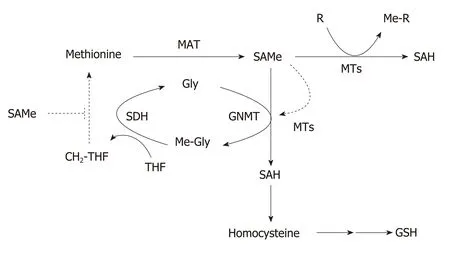
Figure 2 Regulation of hepatic S-adenosylmethionine homeostasis. Hepatic S-adenosylmethionine (SAMe)content is regulated by the concerted activity of methionine adenosyltransferase (MAT) and glycine Nmethyltransferase (GNMT). Methionine is mainly metabolized by the liver where is converted to SAMe by the enzyme MAT using ATP as co-substrate. SAMe, the main cellular methyl donor, is converted to S-adenosylhomocysteine(SAH) by a legion of methyltransferases (MTs) that catalyze the methylation of multiple substrates (DNA, proteins,phospholipids, small molecules, toxic and waist products). Excess SAMe is catabolized by GNMT, the most abundant hepatic MT, to prevent undesirable methylations. The GNMT-sarcosine dehydrogenase (SDH) pathway recycles the excess of methyl groups via generation of methylene-tetrahydrofolate (CH2-THF) and the methylation of homocysteine to regenerate methionine (not shown) to maintain SAMe homeostasis. SAH is converted to homocysteine, a metabolic crossroad that can be used for the regeneration of methionine (not shown) or the synthesis of glutathione depending on whether the concentration of SAMe is low or high, respectively. SAMe is an allosteric activator of GNMT and an inhibitor of the re-synthesis of methionine via the CH2-THF pathway (broken lines). CH2-THF: 5,10-methylene-tetrahydrofolate; Gly: Glycine; GNMT: Glycine N-methyltransferase; MAT:Methionine adenosyltransferase; Me-Gly: Methylglycine (sarcosine); Me-R: Methylated product; MTs:Methyltransferases; MTHF: 5-methyltetrahydrofolate; R: Methylation substrate; SAH: S-adenosylhomocysteine;SAMe: S-adenosylmethionine; SDH: Sarcosine dehydrogenase; THF: Tetrahydrofolate.
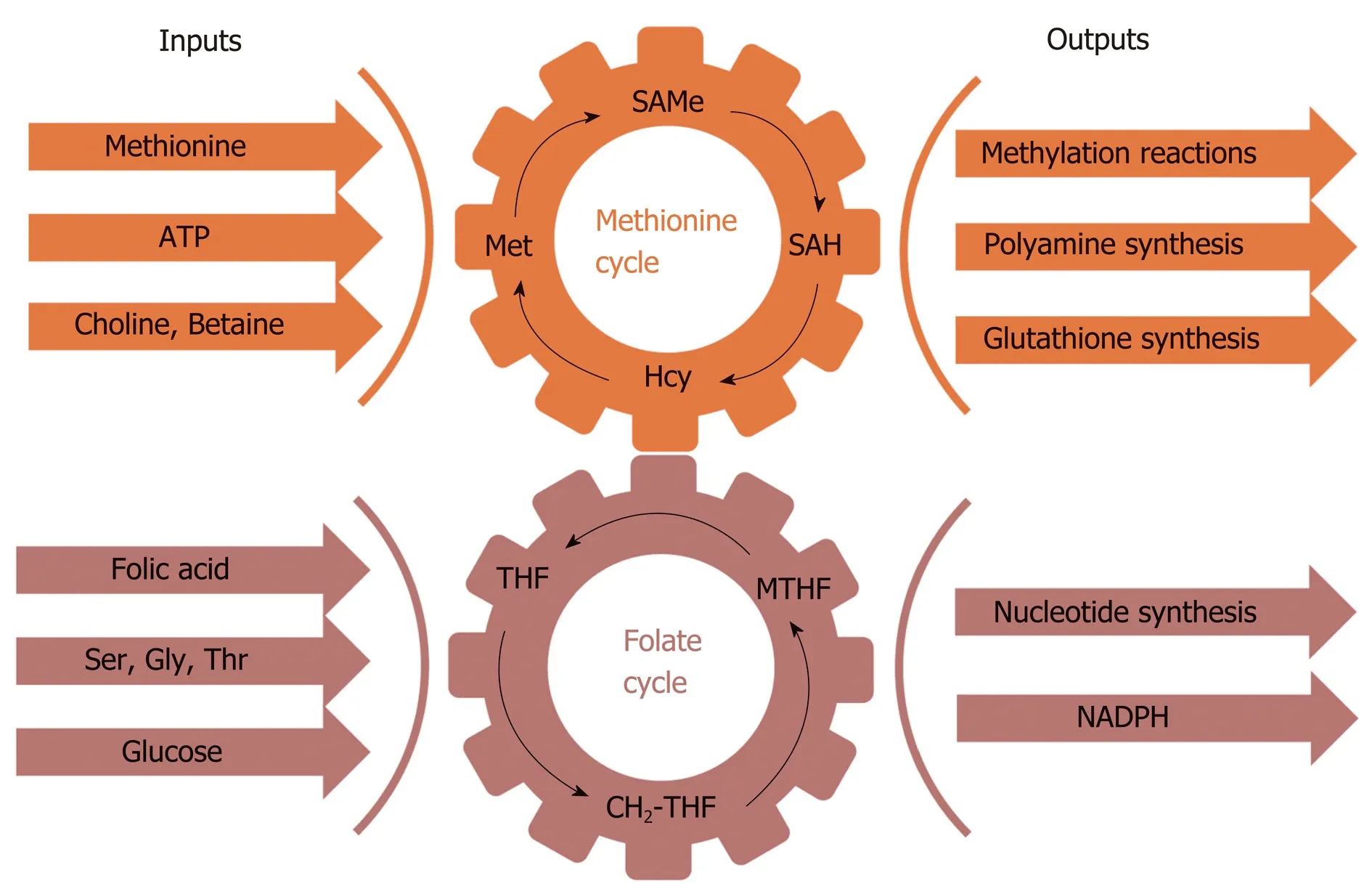
Figure 3 Schematic representation of one carbon metabolism. One carbon metabolism involves multiple physiological processes in which one carbon units circulate from different nutritional and amino acids inputs (choline,betaine, folic acid, glucose, methionine, serine, glycine and threonine), mediated by S-adenosylmethionine and 5-methyltetrahydrofolate, and are converted into a wide variety of outputs, such as the methylation of phospholipids,protein and DNA, and the synthesis of glutathione, polyamines, nucleotides, and reduced nicotinamide adenine dinucleotide phosphate. CH2-THF: Methylene tetrahydrofolate; Gly: Glycine; GSH: Glutathione; Hcy: Homocysteine;Met: Methionine; MTHF: 5-Methyltetrahydrofolate; NAPDH: Reduced nicotinamide adenine dinucleotide phosphate;SAH: S-denosylhomocysteine; SAMe: S-adenosylmethionine; Ser: Serine; THF: Tetrahydrofolate; Thr: Threonine.
IDENTIFICATION OF NAFLD SUBTYPES
Despite the current essential role of biopsy for NAFLD diagnosis, its use as a tool for determining the different metabolic pathways that lead to the initiation and progression of NAFLD is certainly limited. Recently, lipidomics has permitted the classification of NAFLD patients into different subtypes corresponding to the main alteration(s) leading, in each case, to the initiation and progression of NASH based on the identification of specific lipid signatures. We identified a unique serum metabolomic profile that distinguished between Mat1a KO and wild type (WT) mice and observed, using a large cohort of 535 serum samples from biopsied NAFLD patients, that nearly half of them showed this Mat1a KO-type (M-subtype)metabolomic signature[37](Table 1). Although classification based on this approach is not indicative of disease progression (M-subtype is equally distributed among patients with steatosis and NASH), a small group of serum metabolites that could differentiate simple steatosis from NASH in the Mat1a KO and in NAFLD patients was also identified. This work defined, for the first time, the metabolic landscape affected by a chronically reduced hepatic SAMe level and demonstrated key abnormalities that were corrected by SAMe treatment, which led to resolution of NASH.
The MCD diet model is a widely-used murine model of NASH but animals lose weight rapidly, have low serum TG levels, and do not become insulin resistant[66]. The addition of 0.1% methionine (normal diet contains 0.3% methionine) minimizes weight loss and yet mice fed the 0.1MCD diet have low liver SAMe content and developed steatosis, inflammation and fibrosis[59]. The mechanism for steatosis included impaired VLDL-TG secretion and reduced GSH, due to the decrease in SAMe content, the concomitant reduction in the synthesis of PC-PUFA through the PEMT pathway, and increased uptake of FA via CD36. Despite the existence of important differences between both models [(1) The protein content of SCD1 is increased in Mat1a KO and decreased in 0.1MCD; and (2) Mitochondrial FA βoxidation is decreased in Mat1a KO and increased in 0.1MCD], the reduction in hepatic SAMe content is the common driver of NAFLD initiation and progression to NASH in both of them and, accordingly, NAFLD patients classified as M-subtype were found to have a metabolic profile similar to the 0.1MCD model[59](Table 1).Treatment of the 0.1MCD mice for two weeks, after the onset of NAFLD, with the SCD1 inhibitor arachidyl amido cholanoic acid (Aramchol, a Phase 2b test drug candidate in a clinical trial for NASH)[67], improved the liver histology[59](Table 1).Aramchol has been shown to improve the three key pathologies associated to NASH:(1) Steatosis, by reducing TG synthesis and increasing VLDL-TG export and FA βoxidation; (2) Inflammation, by decreasing lipotoxicity; and (3) Fibrosis, by downregulation of collagen production by stellate cells[59]. We speculate Mat1a KO mice, and therefore NAFLD patients with M-subtype serum metabolomic profile, will likely benefit from Aramchol treatment.
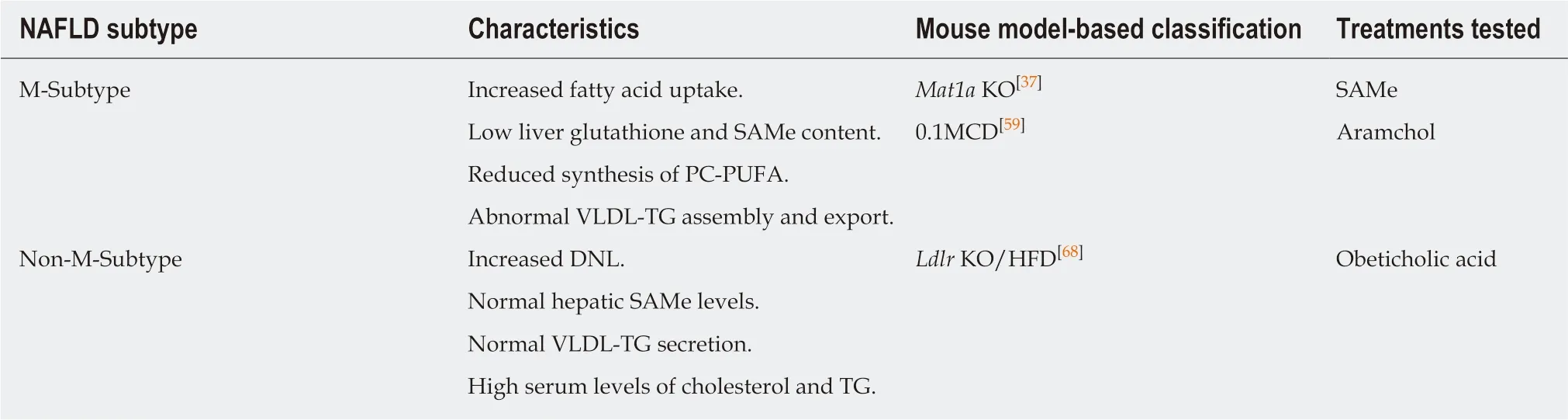
Table 1 Nonalcoholic fatty liver disease subtype classification
Interestingly, nearly all NAFLD patients classified as having a non-M-subtype,according to both the Mat1a KO and 0.1MCD metabolomics models of NASH, were found to have a lipidomic signature similar to that found in low-density lipoprotein receptor (Ldlr) KO mice fed a high fat diet (HFD)[68](Table 1). This mouse model (Ldlr KO/HFD) shows high serum levels of cholesterol and TG, normal liver SAMe, and develop NASH and fibrosis. Treatment of the Ldlr KO/HFD mice for ten weeks, after the onset of NAFLD, with the Farnesoid X Receptor agonist Obeticholic acid (OCA, a Phase 3 test drug candidate in a clinical trial for NASH)[69], nearly normalized the liver histology, reduced blood ALT, AST and TG levels and tended to lower cholesterol content[68]. It would be interesting to determine if in the Aramchol and OCA clinical trials for NASH, patients that responded to treatment were enriched in M- and non-M-subtype, respectively.
However, this approach also results in a certain number of unclassified patients(named as indeterminate)[37,59], which can be inherently linked to the unsupervised classification methodology and validation procedure. Potential integration of other omics data as well as clinical parameters may improve this novel subtyping approach of NAFLD patients, allowing further interpretation of the complex biochemical processes and the heterogeneity of the disease.
CONCLUSION
To understand the pathogenesis of NASH, a useful conceptual framework is that the liver's capacity to accumulate and export TG supports two crucial physiological functions (1) Storing highly energetic, but also highly cytotoxic, FA stably as TG, and(2) Placing into circulation the right amount of VLDL-TG to meet the energy needs of extrahepatic tissues. Both functions collide when the IHTG content exceeds 5% and hepatocytes must safely handle and accumulate excess FA into TG without increasing the rate of VLDL-TG export[13,14]. The maximum capacity to safely handle FA by the liver in the presence of increasing levels of IHTG may vary between individuals depending on the variable contributions from different molecular pathway(s) that result in TG accumulation; and NASH may develop when this maximum capacity is exceeded. The observation that it is possible for NAFLD patients to develop NASH at various grades of steatosis, supports this notion. However, clinical trials currently designed for the treatment of NASH are based on the mechanism of action of a drug that is administered to patients without confirming if that specific molecular pathway is altered; which is against the view that NASH pathogenesis has diverse drivers.Understandably, no more than 40% of patients in these trials have shown a positive response to treatment[26]. Alternatively, a comprehensive landscape of the main NASH drivers may be obtained, for example, by integrating multiomics data of well-defined mouse models of NASH, for which the efficacy of different drugs have been validated, with the multiomics data of a large cohort of well-characterized NASH patients following a similar procedure to that previously described[37]. Such a strategy would associate patients' multiomics signatures to specific therapies that could be validated reanalyzing the data of clinical trials where the efficacy of these drugs has been tested. In addition, this approach may allow advances in our understanding of the complex biochemical processes and pathophysiological responses in NAFLD[70,71].Moreover, it will be also important to integrate gene products, mRNA, proteins and metabolites, with environmental factors, such as diet and life style[72,73]. Finally, this strategy may be extended to the identification of optimal therapeutic drug combinations.
杂志排行

World Journal of Gastroenterology的其它文章
- Immunotherapy for hepatocelluiar carcinoma:Current and futrure
- Application of Big Data analysis in gastrointestinal research
- Imaging biomarkers for the treatment of esophageal cancer
- Development and in vitro study of a bi=specific magnetic resonance imaging molecular probe for hepatocellular carcinoma
- Effect of NLRC5 on activation and reversion of hepatic stellate cells by regulating the nuclear factor-κB signaling pathway
- Freeze-dried Si-Ni-San powder can ameliorate high fat diet-induced non-alcoholic fatty liver disease