固相萃取-超高效液相色谱串联质谱法同时测定保健品中5种生物碱
2019-07-09汪晨霞江蓝蓝寻知庆郭新东黄金凤
王 强,汪晨霞*,江蓝蓝,寻知庆,郭新东,黄金凤
(广州质量监督检测研究院,广东 广州 511447)
生物碱(alkaloids)是一类存在于多种植物体内,由植物次生代谢产生的含氮有机化合物,种类繁多,具有显著的药理活性[1],如阿托品为抗胆碱药,可用于镇静、止痛;茶碱具有抗炎、抗过敏的作用;毛果芸香碱与毒扁豆碱均为胆碱酯酶抑制药;同时它们也对生物机体具有毒性,会引起恶心、呕吐、心悸等症状,严重时可能对神经系统、消化系统等造成损害导致死亡。如阿托品可能会引起中枢神经毒性[2];烟碱会引起植物神经系统麻痹;毛果芸香碱和毒扁豆碱会引起恶心、腹痛等不良反应;茶碱中毒会引起心律失常、昏迷等症状。
保健品是一类具有营养性、食品性天然药品性质,能调节人体机能的功能性食品。近年来,随着人们生活水平的提高,大家越来越关注自身的健康状况,使得保健品具有极其广阔的市场前景,但有一些不法商家在利益驱使下一味追求功效,向保健品中非法添加药物,如在减肥产品中非法添加麻黄碱或茶碱[3-4];添加阿托品用于缓解胃溃疡[5]等,这些所谓的保健品虽然功效显著,但若使用不当,将对消费者的健康和生命造成威胁,因此,人们在关注保健品显著保健功效的同时,其安全性问题也引起了广泛关注。国家食品药品监督管理总局于2012年发布了《保健食品中可能非法添加的物质名单(第一批)》[6],其中就有麻黄碱;卫生部印发的《保健食品禁用物品名单》[7]明确列出中草药颠茄,其抗胆碱功效就来自于植物体内存在的阿托品、颠茄碱以及东莨菪碱等生物碱。但目前针对保健品中生物碱的检验方法缺失严重,生物碱检测方面的研究主要集中在体液[8]、植物饮料[9]和中草药[10]等几个领域,由于保健品成分复杂,这些方法并不能完全适用于保健品中生物碱的检测,因此,建立一个高效灵敏的针对保健品中生物碱的检测方法对于严格控制保健品质量,保证消费者的健康安全具有非常重要的研究意义。
目前,应用于生物碱检测的主要方法包括毛细管电泳法(capillary electrophoresis,CE)[11]、薄层色谱法(thin layer chromatography,TLC)[12]、高效液相色谱法(high performance liquid chromatography,HPLC)[13]、气相色谱-串联质谱法(gas chromatography-tandem mass spectrometry,GC-MS/MS)[14]和高效液相色谱-串联质谱法(high performance liquid chromatography-tandem mass spectrometry,HPLC-MS/MS)[15-20]等,如LIN A H等[21]通过LC-MS/MS测定了拳血浆中5种雷公藤多苷生物碱的含量以及研究其药代动力学行为;姜昕易等[22]建立了一种HPLC同时测定黄柏中5种生物碱含量的方法。生物碱类化合物大部分沸点高、气化难,适用于用液相色谱法进行分离,在HPLC的基础上发展起来的超高效液相色谱法(ultra-high performance liquid chromatography,UPLC)仪器性能更优、检测速度更快;电喷雾质谱技术是目前应用最广泛的“软电离”质谱技术,具有灵敏度高、专属性强、操作简便的优点,因此,选用超高效液相色谱-电喷雾串联质谱(ultra-high performance liquid chromatographyelectrospray ionization-tandem mass spectrometry,UPLC-ESIMS/MS)可更好地满足保健品中生物碱分析检测的高要求。本研究采用超声快速提取样品、固相萃取(solid phase extraction,SPE)技术净化,建立了一种同时测定保健品中5种生物碱(烟碱、毛果芸香碱、茶碱、毒扁豆碱、阿托品)的UPLC-MS/MS方法,5种生物碱的结构式如下:
1 材料与方法
1.1 材料与试剂
烟碱(CAS:54-11-5)、硫酸阿托品(CAS:55-48-1)、茶碱(CAS:58-55-9)、毒扁豆碱(CAS:57-47-6):上海Anpel实验科技股份有限公司;毛果芸香碱(CAS:92-13-7):美国Pharmacopeical Convention公司;所有标准品的纯度均>98%。乙腈、甲酸、氨水、甲醇(均为色谱级):美国Merck公司;HLB固相萃取柱(500 mg、6 mL)、乙酸铵:上海Anpel实验科技股份有限公司;Cleanert PCX(500 mg、6 mL)和Cleanert PEP(500 mg、6 mL):天津Agela Technologies公司。
1.2 仪器与设备
Thermo Ultimate 3000高效液相色谱仪、TSQ Endura三重四级杆质谱仪:美国Thermo公司;Milli-Q超纯水器:美国Millipore公司;MS3涡旋仪:德国IKA公司;KQ-250DV数控超声波清洗器:昆山超声仪器有限公司;3K15离心机:德国Sigma公司;TurboVap LV氮吹仪:美国Caliper公司。
1.3 方法
1.3.1 标准溶液的配制
混合标准中间液:分别称取适量5种生物碱标准品用乙腈配制成单标储备液(1 000μg/mL);然后分别吸取适量的单标储备液,用乙腈稀释成质量浓度均为20μg/mL的混合标准溶液。再用该标准溶液稀释成质量浓度为100μg/L混合标准中间液。
混合标准工作液:吸取适量的混合标准中间液用空白基质提取液稀释定容,得到质量浓度分别为0.2μg/L、0.5μg/L、1.0μg/L、5.0μg/L、10μg/L、20μg/L的系列混合标准工作溶液。所有标准溶液均需于4℃冰箱里保存。
1.3.2 样品制备及前处理
片剂、胶囊:取20片/粒样品置于粉碎机中,研磨均匀成粉末状,室温保存。称取0.5 g(精确至0.001 g)于10 mL具塞比色管中,边涡旋边加入4 mL 0.1%甲酸水-乙腈(3∶1,V/V)将样品混匀,超声(1 000 W、40 kHz)萃取20 min,待样品温度降至室温时用0.1%甲酸水-乙腈(3∶1,V/V)定容至5.0 mL。吸取适量提取液于高速离心管中,10 000 r/min离心5 min,取1.0 mL上清液待净化。
1.3.3 样品净化
Cleanert PEP固相萃取柱依次用5 mL甲醇和5 mL水活化待用;取1.0 mL上清液上样,待样品全部通过小柱用5 mL水淋洗小柱,最后用5 mL甲醇洗脱样品,收集全部洗脱液,50℃氮吹至近干,用0.1%甲酸水-乙腈(9∶1,V/V)定容至1.0 mL,涡旋混匀,过0.22μm滤膜待UPLC-MS/MS分析。
1.3.4 检测条件
超高效液相色谱条件:Waters ACQUITY HSS T3液相色谱柱(2.1 mm×100 mm×1.8μm);流动相:A为0.1%甲酸水(含10 mmol/L乙酸铵),B为0.1%甲酸乙腈,梯度洗脱程序:0~1 min,90%A;1~4 min,90%~50%A;4~5 min,50%~90%A;5~6 min,90%A;柱温30℃;进样量5μL;流速0.3 mL/min。
质谱条件:电喷雾离子源(electrospray ionization source,ESI),正模式;数据采集:多反应监测(multireaction monitoring,MRM);电喷雾电压3 500 V;离子化温度300℃,辅助气流速12 L/min;喷雾器温度350℃,鞘气流速40 L/min;碰撞气为氩气,1.5 mTorr;各组分的出峰时间、质谱信息、碰撞能压及射频电压见表1。

表1 5种生物碱的质谱参数Table 1 MS parameters of five alkaloids
2 结果与分析
2.1 检测条件的优化
2.1.1 质谱条件的优化
采用TSQ Endura三重四级杆质谱仪,在多反应监测模式下,对5种生物碱的质谱条件进行优化。将5种生物碱单标标准溶液,在ESI+和ESI-两种模式下进行扫描。结果得出,5种生物碱易在ESI+模式下得到[M+H]+准分子离子峰,质谱信号响应较强。可能因为生物碱的N原子上含有孤对电子,容易加H+形成正电荷离子,然后选择该特征准分子离子峰为母离子进行二级质谱分析,以响应值最大的碎片离子定为定量离子,次级响应最大的碎片离子定为定性离子,优化射频电压及碰撞能等质谱参数。最佳质谱条件如表1,在此条件下5种生物碱的MRM色谱图如图1所示。

图1 5种生物碱的多反应监测色谱图Fig.1 Multiple reaction monitoring chromatogram of five alkaloids
2.1.2 色谱柱的选择
本次实验考察了WatersACQUITYUPLCBEH(2.1 mm×100 mm×1.7 μm)、Waters ACQUITY HSS T3(2.1 mm×100 mm×1.8μm)和Agilent Eclipse Plus C18(2.1 mm×100 mm×1.8μm)三种液相色谱柱对5种生物碱的分离效果。结果发现,Agilent Eclipse Plus C18和Waters ACQUITY UPLC BEH对这5种生物碱保留能力较差,毛果芸香碱在Agilent Eclipse Plus C18柱上峰形分叉,毒扁豆碱在Waters ACQUITY UPLC BEH柱上拖尾严重;而所有目标化合物在Waters ACQUITY HSS T3柱上均能实现有效分离、保留效果好。这是因为ACQUITY UPLC HSS T3采用高强度硅胶基质,特有的C18烷基键合以及有效的端基封尾技术,可以兼容纯水流动相,并提高了对水溶性好、极性大的化合物的保留能力。因此,本实验选用Waters ACQUITY HSS T3分析生物碱类化合物,可改善目标化合物峰形以及保留稳定性。
2.1.3 流动相的优化
在电喷雾质谱正离子模式下流动相中加入酸性介质,有利于被测组分的离子化,从而提高其质谱响应和检测灵敏度。本实验分别考察了甲醇-0.1%甲酸(V/V)、乙腈-0.1%甲酸(V/V)、甲醇-0.1%三氟乙酸(V/V)和乙腈-0.1%三氟乙酸(V/V)四种混合溶剂体系,包括两种有机相、两种酸性介质作为流动相对5种生物碱的色谱分离效果和离子化程度的影响。结果表明,有机相为甲醇时质谱响应值低且保留重现性差;水相为0.1%三氟乙酸时茶碱和毛果芸香碱的响应比0.1%甲酸的低50%。流动相为乙腈-0.1%甲酸水(V/V)时,色谱信号响应高但目标物峰形拖尾严重、有的化合物会出现峰分叉现象,色谱分离效率低。这可能是因为生物碱这一类碱性化合物的阴离子会与反相液相色谱柱固定相上裸露的硅胶基质相互作用,从而导致该类化合物在柱上保留过强、峰形拖尾,增加了分离难度,可以通过在流动相中加入乙酸铵可以改善生物碱类化合物的峰形和分离效果[22]。将10 mmol/L乙酸铵中加入0.1%甲酸水溶液,实验表现出明显的改善效果:峰展宽变小,峰拖尾现象得到明显改善,峰形尖锐、色谱分离度得到了提高,这可能是因为乙酸铵防止了固定相中硅醇活性基团与目标物组分相互作用。因此,本实验采用乙腈与0.1%甲酸水(含10 mmol/L乙酸铵)(V/V)混合的溶液作流动相进行梯度洗脱。
2.2 实验方法的优化
2.2.1 样品提取溶剂
目标物的极性差异较大,因此,适合采用混合溶剂来进行分离提取。生物碱通常有两种提取方式:一种是酸水提取法,大部分生物碱一般以盐的形式存在,用酸水提取使得生物碱酸化,最大程度转化成盐增大其在水中的溶解度;第二种是碱水提取法,用碱水碱化可将生物碱游离出来,再用有机溶剂提取。由于酸化前处理操作简单,因此,选用0.1%甲酸水-乙腈(V/V)混合溶剂对样品进行提取,结果见图2。

图2 不同提取溶剂对回收率的影响Fig.2 Effect of different extraction solvents on recovery rates
由图2可知,5种生物碱随着混合溶剂体系中水相的增大,提取回收率在逐渐增加,这说明5种生物碱在0.1%甲酸的作用下酸化效果好,水溶性增大,增强了提取效率。因此,选用0.1%甲酸水∶乙腈=3∶1(V/V)为保健品中目标物的提取溶剂。
2.2.2 样品提取方式
保健品的形态多样,有口服液、片剂、胶囊等。片剂与胶囊样品在提取溶剂中不易分散且保健品前处理所用的样品量和提取溶剂量都较少,而超声萃取法是利用超声波的空化作用、机械效应和热效应等加速样品中各组分的释放、帮助其扩散和溶解于提取溶剂中,可以显著改善提取效果的一种有效的提取方法。因此,适合用于保健品的前处理过程,操作简单,可帮助样品分散均匀、缩短提取时间,提高提取效率。本实验研究了不同超声时间对保健品中目标物提取效率的影响,结果见图3。

图3 不同超声时间对回收率的影响Fig.3 Effect of different ultrasonic time on recovery rates
由图3可知,随着超声时间的延长,提取溶剂与样品接触的越充分,目标物的提取回收率逐渐增加,到20 min后开始变化不大,这说明超声时间>20 min后,样品的萃取过程趋于一定的平衡,从节约时间和能源的角度考虑,选择提取效果已经能满足分析要求的20 min为实验的超声时间。
2.2.3 样品净化方法
由于保健品基质复杂、干扰物多,若超声萃取后直接进行分析检测,基体干扰大。因此,需要根据被测目标物的性质,选择合适的净化方式,排除基体干扰,分离和富集样品中的目标物。固相萃取法利用选择性吸附与选择性洗脱的色谱分离原理,以达到富集、分离、净化的目的,可用于降低样品基质干扰、提高目标物的选择性和检测的灵敏度,在食品、化工等领域得到了越来越广泛的应用。常用的萃取柱包括HLB、C18、NH2不同属性的填料等,由于生物碱极性较大,不适合用反相固相萃取净化;而PEP和HLB的填料均为一类两由种不同性质的化合物组合而成的共聚物,可同时表现出亲水亲酯的特性,从而对极性和非极性化合物具有均衡的吸附作用;MCX作为阳离子交换柱,适合于离子型化合物的分析。因此,本研究分别考察了MCX、PEP和HLB固相萃取柱的净化效果,结果见图4。
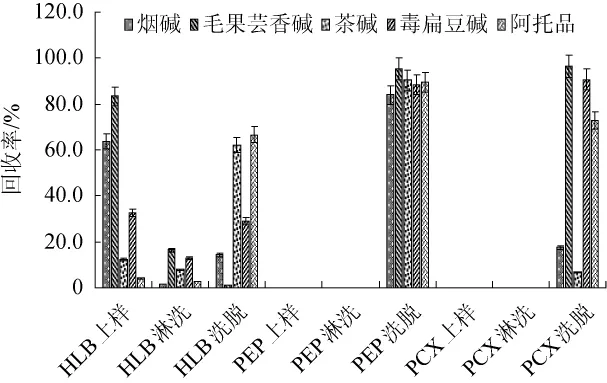
图4 不同固相萃取柱对回收率的影响Fig.4 Effect of different solid phase extraction columns on recovery rates
由图4可知,加标样品提取液上样后,Cleanert PEP和Oasis HLB先用5 mL水淋洗再用5 mL甲醇洗脱,Cleanert PCX用5 mL甲醇淋洗再用5 mL 5%氨化甲醇(V/V)洗脱。结果表明,PEP柱对所有目标物能够实现有效的分离富集;HLB对这5种生物碱的保留较差,目标物分别在上样、淋洗和洗脱的过程中均有流出;MCX柱对毛果芸香碱、毒扁豆碱和阿托品的的回收效果较好,但阿托品的回收率稳定性差,可能是因为阿托品在用氨化甲醇洗脱时碱性条件下发生了水解,而烟碱和茶碱的回收率较低可能因为这两种化合物碱性较强,洗脱所用的氨化甲醇碱性不足以将其置换下来。因此,本实验选择PEP固相萃取小柱净化。
2.3 方法学技术指标考察
2.3.1 基质效应
基质效应是指样品中除分析物以外的组分会对分析物的分析产生明显的干扰,影响分析结果的准确性。在UPLCMS/MS中共流出的基质会与分析物在离子化过程中产生作用而影响目标物的离子化效率。所以基质效应可能会影响检测方法的灵敏度、精密度和准确度。而不同类型的保健品,基质效应也存在一定的差异。本实验采用空白基质提取液配制标准工作液,空白基质溶液的制备与样品前处理过程一致,这样标准工作溶液和样品溶液都具有相似的基质环境,从而确保定量所用线性方程的适用性。
2.3.2 线性范围及方法检出限
在最佳UPLC-MS/MS仪器条件下测定0.2~20μg/L系列标准工作混合溶液,以目标物的质量浓度(x)为横坐标,质谱响应(y)为纵坐标,得到线性回归方程。以0.2μg/L标准工作混合溶液的信噪比(S/N=3)结合样品前处理过程计算得出方法的检出限(limit of detection,LOD),其线性方程、相关系数和方法检出限见表2。由表2可知,在0.2~20μg/L范围内5种生物碱线性回归方程相关系数R2均≥0.999 0,方法检出限为0.6~0.8 μg/kg。
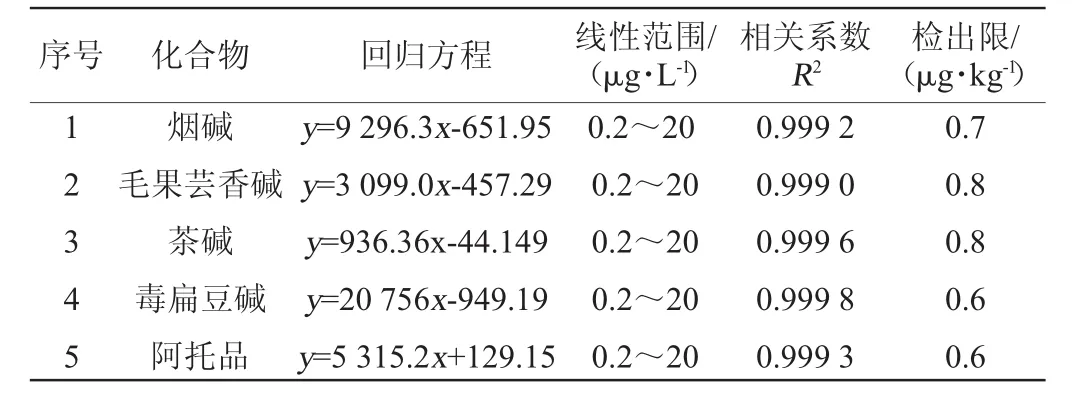
表2 5种生物碱的线性方程、线性范围、相关系数及检出限Table 2 Linear regression equations,linear range,correlation coefficients and limit of detection of five alkaloids
2.3.3 方法精密度及回收率
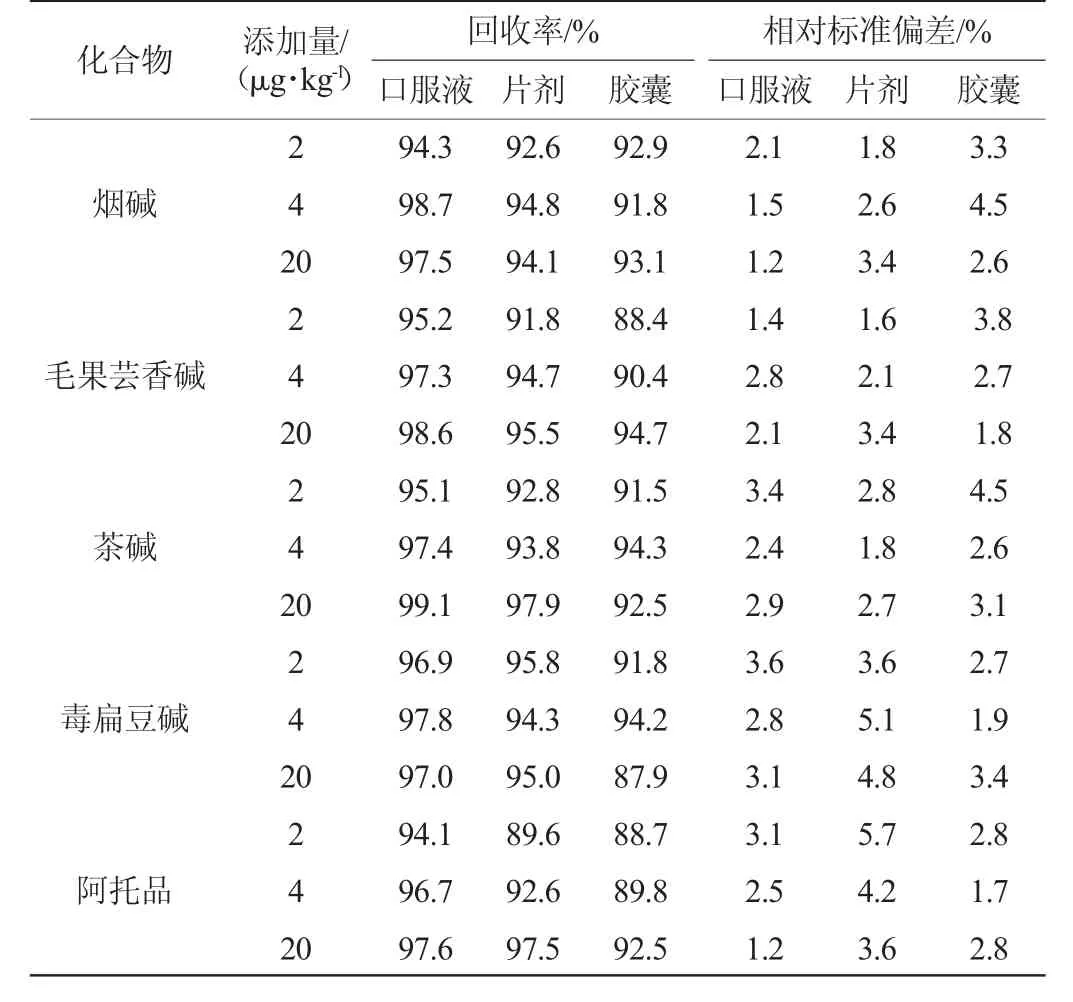
表3 5种生物碱加标回收率及精密度实验结果Table 3 Results of standard recovery rate and precisions tests of five alkaloids
在最优的实验条件下,选取胶囊、口服液、片剂3种基质的空白样品进行添加回收实验,分别添加3个加标浓度,每个加标浓度平行测定6次,计算回收率及其相对标准偏差,结果见表3。由表3可知,3个添加浓度的回收率为87.9%~99.1%,相对标准偏差(relative standard deviation,RSD)为1.2%~5.7%。结果表明,该方法精密度和准确度良好。
2.4 实际样品测定
随机购买了本市市售的口服液、片剂和胶囊共30种不同类型的保健品采用本方法进行检测,均未检出5种生物碱。
3 结论
本研究选用超高效液相色谱-串联质谱联用技术,针对保健品中可能出现的非法添加成分,建立同时测定5种生物碱(烟碱、毛果芸香碱、茶碱、毒扁豆碱、阿托品)的方法。通过优化色谱条件和质谱参数得到了最优的检测条件;优化样品提取时间及固相萃取净化方法对目标物实现了较好的提取和富集。该方法样品前处理操作简单;对这几种生物碱的检测线性范围宽、灵敏度高、检出限低;所测结果准确度高、重现性好,可为保健品的质量安全监管提供技术支持。
