食品中滑石粉含量测定样品前处理方法的改进
2019-07-09杨庆文王争争杨宇霞
杨庆文,王争争,赵 欣,李 慧,杨宇霞
(山西省生物研究院有限公司,山西 太原 030006)
滑石粉是将滑石经去净泥土、杂石,再经粉碎、浮选、干燥而制成[1],主要成分为天然的水合硅酸镁,其分子式为Mg3Si4O10(OH)2[2]。滑石粉的种类可分为化工级、陶瓷级、化妆品级、医药食品级、造纸级和水镁石粉等[3-6]。根据国标GB 2760—2014《食品安全国家标准食品添加剂使用标准》[7]中的有关规定,滑石粉可以作为食品添加剂在加工环节中应用于凉果类、话化类食品的生产,其最大使用限量为20.0 g/kg。医学研究表明,人体长期大剂量服用滑石粉会发生肾硅酸盐结石[8],肾功能不全者可能会出现眩晕、昏厥、心律失常以及异常疲乏无力等现象。另外,工业级滑石粉中含有一定量的伴生杂质石棉和石棉纤维[9],这些异物进入人体内会附着、沉积在肺部,易诱发石棉肺、胸膜间皮瘤、肺癌等肺部疾病。2017年10月27日,世界卫生组织国际癌症研究所机构公布将石棉或含石棉状纤维的滑石粉列入3类致癌物清单中[10-11]。目前,市场上有部分不法厂商为了牟取暴利,往往超范围、超限量甚至跨等级地在食品加工生产过程中大量违规使用滑石粉,如有的厂家在面粉中违规添加滑石粉[12-14],行话叫“串粉”,严重的竟达20%以上;有些炒瓜子的商贩为了使炒制出来的瓜子看起来美观、光滑,也往瓜子里加滑石粉。更严重的是,某些黑心商家为了降低成本竟然使用工业级滑石粉添加到食品中去,这些做法严重危害了广大消费者的身体健康,有时甚至会危及生命。由此可见,食品中滑石粉的检测是食品安全检测中非常重要的一环。
过去,食品中滑石粉的检测是通过人的肉眼和感官判别[13]或者通过一些简单的化学方法来测定[14-15],这些方法只适合那些食品中含有大量滑石粉的情况,且只能作定性判别,对滑石粉的掺入量难以衡量。国外则有采用红外光谱法测定地质样品中滑石粉的报道[16],甚至对滑石粉的表面形状及可吸入颗粒进行测定[17-19]。近年来,随着人们不断摸索、探讨,火焰原子吸收光谱(flame atomic absorption spectrometry,FAAS)法已成为测定食品中滑石粉含量的通行办法[20-23]。目前,大都依据国标GB 5009.269—2016《食品安全国家标准食品中滑石粉的测定》[24]测定食品中的滑石粉含量。该标准采用的方法原理是先用硝酸+高氯酸(或硝酸+过氧化氢)消化试样,过滤,将滑石粉与其他含镁物质分离,然后再加氢氟酸反应生成溶于水的镁盐,最后采用火焰原子吸收光谱(FAAS)法测定镁含量,并根据镁含量计算食品中滑石粉的含量。按照此方法前处理,样品消解需经历前后两个消化过程,中间需要进行冷却、过滤、洗涤等操作步骤。多次试验经历显示,整个消解过程一般用时约11 h。消解过程中需要进行称样、加液、移位和准备器皿试剂等,在不考虑加班的情况下受上、下班时间限制需要升温、降温等实际情况,至少需要一个半工作日(实际上往往需要2 d)。该消解方法不仅费时费力,而且在两次消解之间为了过滤进行的一系列操作增加了样品损失或受到污染的机会。
本研究对国标中湿消解法前处理方法进行改进,新的消解方法既缩短了工时,又减轻了工作量,而检测结果是令人满意的,甚至比原方法更好。本研究将为一线检测人员提供一种新的测试面粉中滑石粉含量的简捷方法。
1 材料与方法
1.1 材料与试剂
乙炔气(纯度≥99.999%):山西泰能气体有限公司;镁标准储备溶液(质量浓度500 mg/L):中国计量科学研究院;掺滑石粉的面粉样品:平度良金面粉厂;滑石粉标准品(试验品):阿法埃莎(中国)化学有限公司;硝酸(分析纯):北京化学试剂研究所;氢氟酸、高氯酸(均为分析纯):国药化学试剂有限公司;盐酸(分析纯):北京化工厂;氯化锶(分析纯):天津市光复精细化工研究所。
混合酸的配制:硝酸+高氯酸(4∶1,V/V)。
氯化锶溶液(15 g/L)的配制:称取氯化锶15 g,加入100 mL纯水和45 mL浓盐酸溶解,用纯水稀释至1 000 mL。
镁标准中间液(100 mg/L)的配制:用氯化锶溶液将镁标准储备液稀释成镁标准中间液。
1.2 仪器与设备
AA 800型原子吸收光谱仪:美国PerkinElmer公司;KY-PE型镁空心阴极灯:北京曙光明电子光源仪器有限公司;EG35B型微控电热板:莱博泰克(大连)科技有限公司。
1.3 方法
1.3.1 火焰原子吸收光谱仪工作条件
波长:285.2 nm;狭缝宽度:0.7 nm;灯电流:6.0 mA;空气流量:17 L/min;乙炔流量:2.3 L/min;燃烧头高度:0 mm;检测时燃烧头偏转45°。
1.3.2 国标GB 5009.269—2016试样前处理湿消解法
称样→加酸、升温(约3 h)→冷却(约2 h)→过滤,洗涤(约1 h)→加酸、升温、赶酸、定容(约5 h)
称取面粉试样0.5 g,放入聚四氟乙烯坩埚中,加入15 mL混合酸,盖上盖,电热板上加热消化1~2 h,至消化液无色透明,如果酸液过少时,可适当补加混合酸后继续加热消化。将坩埚取下,冷却,用20 mL纯水冲洗坩埚,将坩埚中的固体残渣完全转移到定量滤纸上,过滤,用100 mL纯水洗滤纸及滤渣。将滤纸及固体残渣放入聚四氟乙烯坩埚中,加入10 mL混合酸和3 mL氢氟酸,盖上盖,电热板上加热消化1~2 h,至消化液无色透明,如果酸液过少时,适当补加混合酸后继续加热消化。待坩埚中的液体接近干时,取下冷却,用氯化锶溶液冲洗坩埚,将消化液转移至100 mL容量瓶中,用氯化锶溶液定容,混匀,该溶液为试样溶液,备用。
试样空白试验与试样消解处理同步进行。用待测试样做空白试验,按照与试样消解处理一样的步骤进行消解处理,只是将滤纸及纸上固体残渣进行消解时不加入氢氟酸,试样中的滑石粉不发生化学反应。试样空白试验的取样量、定容体积和稀释倍数与试样消解处理相同。
1.3.3 改进后的试样消解处理湿消解法
称样→加酸、升温、赶酸、定容(约6 h)
称取面粉试样0.5 g,放入聚四氟乙烯坩埚中,加入15 mL混合酸和3 mL氢氟酸,盖上盖,电热板上加热消化,至消化液无色透明。如果酸液过少时,适当补加混合酸后继续加热消化。待坩埚中的液体接近干时,取下冷却,用氯化锶溶液洗坩埚,将消化液转移至100 mL容量瓶中,用氯化锶溶液定容,混匀、静置,备用。
试样空白试验与试样消解处理同步进行。用待测试样做空白试验,按照与试样消解处理一样的步骤进行消解处理,只是不加氢氟酸,试样中的滑石粉不发生化学反应。
1.3.4 试样加标回收试验
称取试样约0.5 g,添加一定量的滑石粉标准品,即为加标样。加标样的消解处理方法同试样。加标样试样溶液制备完毕后,取1 mL消解液至100 mL容量瓶中,用氯化锶溶液定容至刻度,混匀、静置,备用。
1.3.5 镁标准工作曲线绘制
镁系列标准工作溶液的配制:分别吸取0、0.50 mL、1.00 mL、2.00 mL、4.00 mL、5.00 mL镁标准中间液于100 mL容量瓶中,用氯化锶溶液定容,配制成质量浓度分别为0、0.50 mg/L、1.00 mg/L、2.00 mg/L、4.00 mg/L、5.00 mg/L镁标准工作溶液。
在1.3.1仪器工作条件下,对镁元素系列标准工作溶液进行测定。以吸光度值(y)为纵坐标,以镁元素质量浓度(x)为横坐标,绘制镁标准工作曲线,得到标准曲线回归方程为y=0.033 5x+0.001 2,相关系数R2=0.999 2。
1.3.6 计算公式
(1)国标前处理方法滑石粉含量计算公式
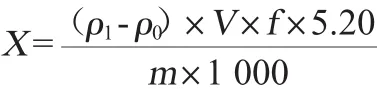
式中:X为试样中滑石粉含量,g/kg;ρ1为试样溶液中镁元素的质量浓度,mg/L;ρ0空白溶液中镁元素的质量浓度,mg/L;V为试样溶液定容体积,mL;f为试样溶液稀释倍数;5.20为镁元素换算为滑石粉(相对分子质量379)的系数;m为试样质量,g;1 000为计量单位换算系数。
(2)改进前处理方法后滑石粉含量计算公式

式中:X为试样中滑石粉含量,g/kg;Mg1为试样中镁元素的含量,g/kg;Mg0为空白试样中镁元素的含量,g/kg;ρ1为试样溶液中镁元素的质量浓度,mg/L;ρ0空白溶液中镁元素的质量浓度,mg/L;V为试样溶液定容体积,mL;f为试样溶液稀释倍数;5.20为镁元素换算为滑石粉(相对分子质量379)的系数;m为试样质量,g;m0为空白试样质量,g;1 000为计量单位换算系数。
2 结果与分析
2.1 分析结果计算
国标GB 5009.269—2016《食品安全国家标准食品中滑石粉的测定》中要求试样空白的取样量要与试样相同。滑石粉含量计算公式按照1.3.6节改变以后,在称取样品时就不必刻意把每一个样品的空白样和平行样都称成相同的质量,这样做的好处是称量样品时较原法更随意,称样速度将大大加快,若样品数量越多优势越明显,可以节约大量时间。当然,样品间的称样量差异也不应太大,以免改变统一的试剂用量。
2.2 两种消解方法的测定结果
为了明确地分析两种消解前处理方法之优劣,进行加标回收试验。在每份约0.5 g样品中分别添加0.050 g、0.100 g、0.150 g滑石粉标准品添加到样品中,加标质量浓度分别约为100 g/kg、200 g/kg、300 g/kg。样品及加标样品消解处理完毕后,再将这些消解液均稀释100倍后上机测试。
根据仪器测定结果,按公式(1)、(2)、(3)计算样品中滑石粉的含量及加标回收试验结果,结果分别见表1、表2及表3。
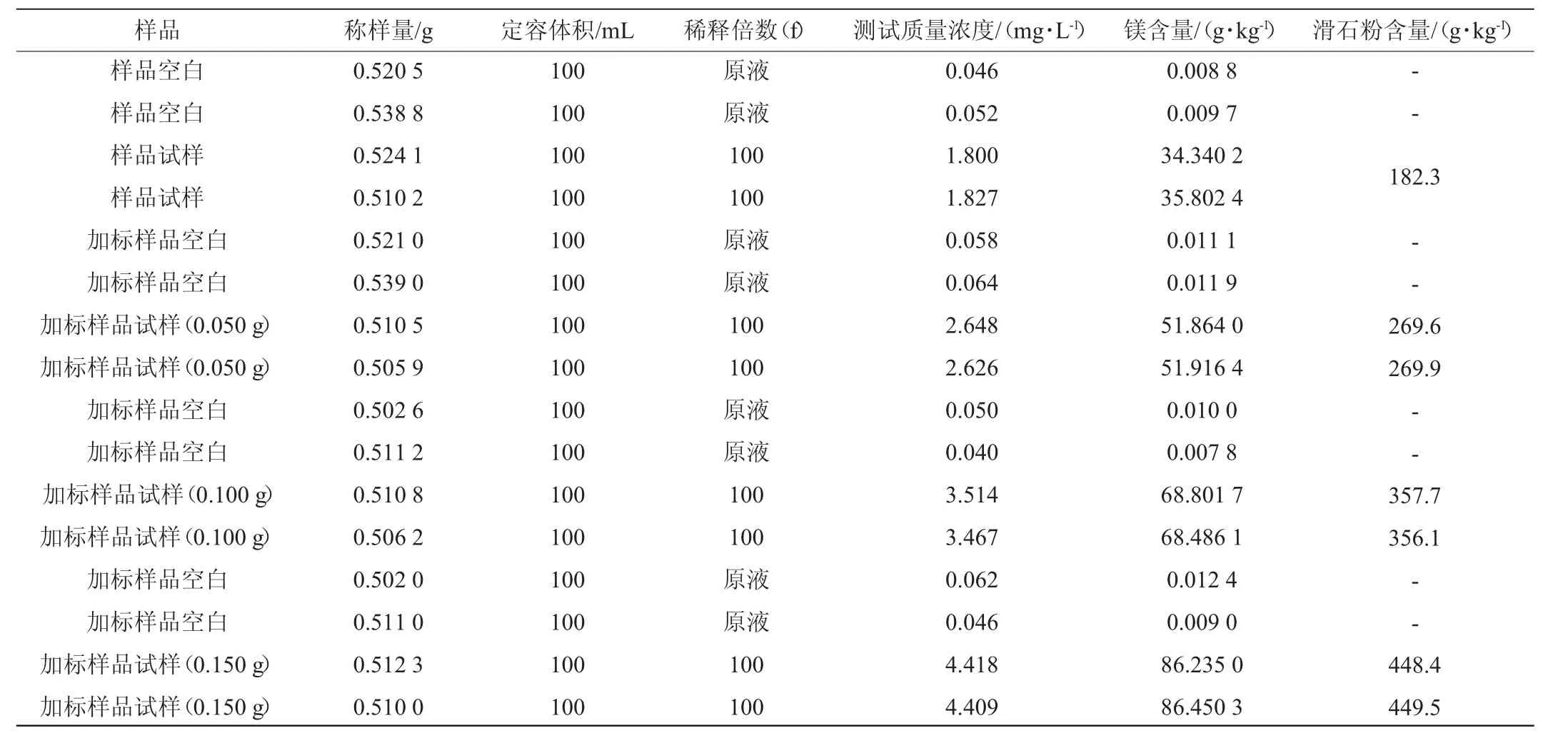
表1 采用国标前处理方法消解样品中滑石粉含量测定结果Table 1 Determination results of talc powder contents in digestion sample by pretreatment method of national standard

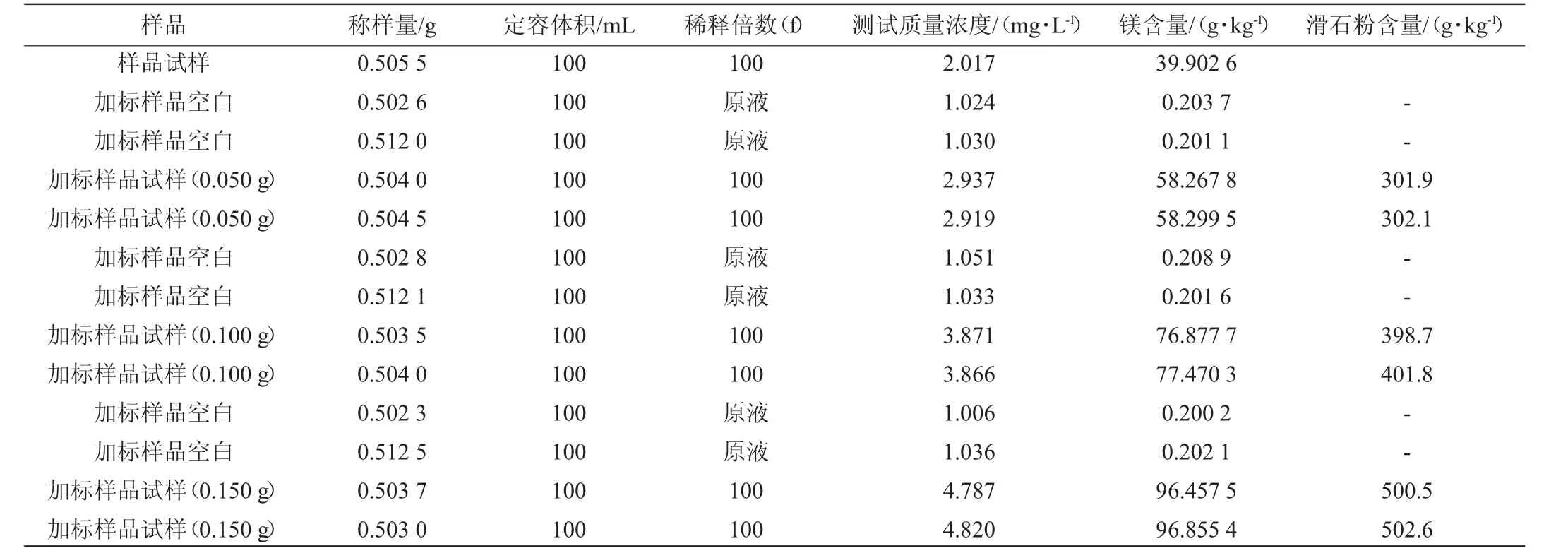
表2 采用改进后前处理方法消解样品中滑石粉含量测定结果Table 2 Determination results of talc powder contents in digestion sample by modified pretreatment method
续表
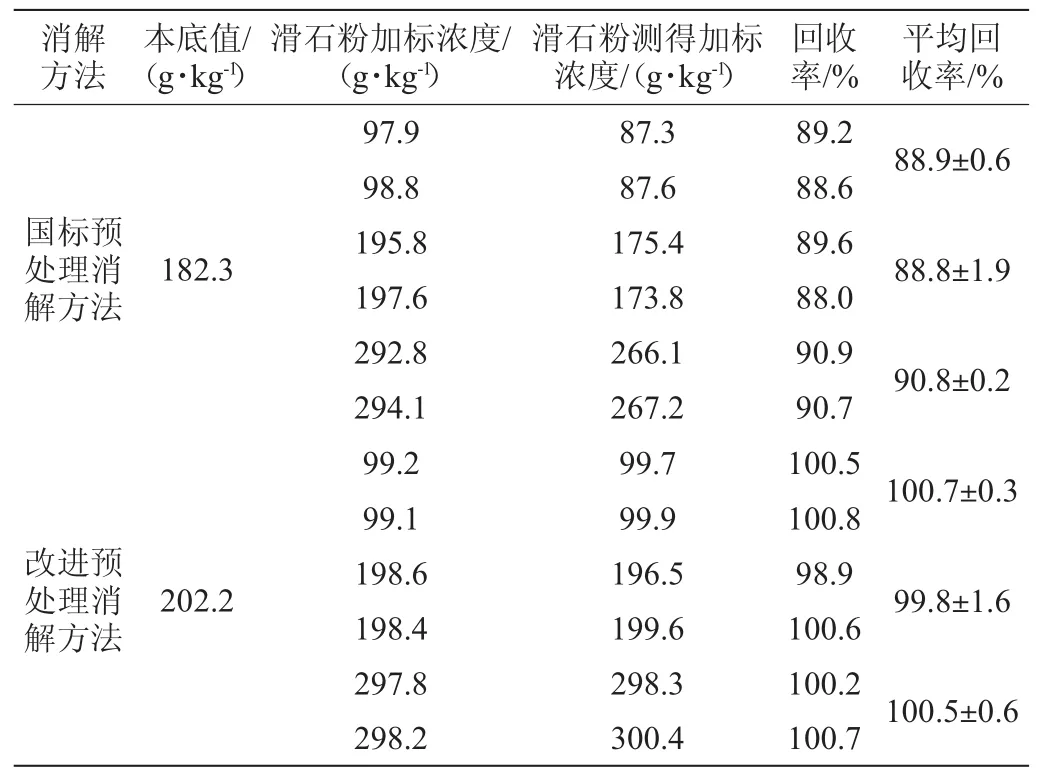
表3 加标回收试验结果Table 3 Results of standard recovery rate tests
由表1~表3可以看出,采用改进后的预处理消解方法得到的结果加标回收率为98.9%~100.8%;国标预处理消解方法得到的结果加标回收率为88.0%~90.9%。结果表明,两种前处理方法得到的结果加标回收率均在80%~120%范围内。因此,对国标预处理消解方法的改进是可行的。
2.3 两种消解方法对比
两种消解方法之间的比较结果见表4。

表4 两种前处理消解方法比较Table 4 Comparison of the two pretreatment digestion methods
由表4可以看出,改进后的消解方法比原消解方法减少了3个操作步骤。每少一个步骤,自然地就会减少试验用时以及样品中待测元素损失或受到污染的机会。在研究试验中,采用新的改进后的消解方法处理样品用时缩短近一半,从使用实验器皿、试剂等耗材角度来算,减少操作程序也减少了器皿、用具和试剂消耗。不难想象,当样品数量较多时,用时、用具及试剂的消耗等将随之大量增加,这时采用改进后的新的消解方法将充分体现出其优势,尤其是在减少检测时间和操作步骤更简单两个方面。
3 结论
与国标前处理消解方法相比,采用改进后的前处理消解方法测定食品中滑石粉时得到的结果更加准确,表明对于原消解法的改进是成功的。采用改进后的消解方法,不仅减少了试剂消耗,简化了操作步骤,更重要的是将极大地缩短检测时间,这对于样品的检测具有非常重要的意义。另外,对计算结果的方法所做的改进使得样品称取不再拘泥,这符合对操作要化繁为简,方便易行的实际要求,而且同样减少了实验用时,故对改进的消解方法是一个良好的补充。从更进一步来看,虽然本法是以湿法消解为基础进行了研究,但基于相同的原理,预测将该法照搬到微波消解法中去也是可行的。
