铁死亡与脑损伤的关系
2019-07-09蒋如如欣综述哈小琴审校
蒋如如,李 欣综述,哈小琴审校
铁死亡是近年来发现的一种由铁依赖的氧化损伤引起的细胞死亡模式,与凋亡、坏死、自噬不同,铁死亡的特征主要是细胞体积缩小和线粒体膜密度增加,没有典型的细胞凋亡和坏死表现[1]。铁死亡作为非凋亡形式的细胞死亡于2012年被首次报道,这一过程最初是用合成的小分子化合物在癌细胞研究中发现的,但后来报道表明它与脑和其他组织急性损伤的细胞死亡事件有关。铁依赖的细胞死亡并不是一个新概念[2],在神经退行性疾病和脑损伤研究中均有与铁和氧化应激的病理细胞死亡事件的诸多描述[3,4],这一现象可通过诱导铁死亡来解释。研究表明,铁死亡涉及人类疾病,在神经退行性疾病和脑损伤研究中发现许多含有与铁和/或氧化应激的病理细胞死亡事件的描述[3],因此,了解铁死亡通路以及其诱因可能会产生治疗脑损伤的新方法。
1 铁死亡定义
铁是地壳中第四大常见元素,它在人体中起着举足轻重的作用。铁对细胞存活至关重要,它参与有氧运输、DNA和ATP合成以及三羧酸(tricarboxylic acid,TCA)循环和电子传递链中各种蛋白质的辅助因子[5]。铁,特别是二价铁的存在,可大大加速人体内饱和脂肪酸的脂质过氧化,铁参与线粒体氧化磷酸化过程、细胞产生活性氧(reactive oxygen species,ROS)以及ATP的产生,铁沉积所引起的氧化反应若超过细胞的抗氧化能力水平则可导致细胞内发生氧化应激反应,直接或间接地引起大分子的物质如蛋白质、核酸和脂质损伤,导致细胞损伤或死亡。这种新发现的细胞死亡形式称为铁死亡。铁死亡与传统意义上的细胞凋亡和坏死不同,是铁依赖性脂质过氧化物的积累所引起的。铁死亡不仅存在于癌细胞中,还存在于神经系统疾病模型系统中的死亡神经元中[6,7],研究发现铁螯合剂去铁胺(deferoxamine,DFO)在保护脑细胞免于死亡方面具有巨大潜力[8]。
2 铁死亡机制
2.1 抑制系统XC-引发铁死亡 系统XC-是膜Na+依赖性半胱氨酸-谷氨酸交换转运蛋白,其是由轻链亚基(xCT,SLC7A11)和重链亚基(CD98hc,SLC3A2)36组成的由二硫键连接起来的异二聚体。当系统XC-将细胞内谷氨酸转运到细胞外的同时将细胞外胱氨酸转运到细胞内,然后胱氨酸在细胞内转化为半胱氨酸以进行谷胱甘肽(glutathione,GSH)的合成,该转运周期与ATP无关,由高浓度的细胞内谷氨酸驱动(见图1)。这使得系统XC-对细胞外谷氨酸的浓度异常敏感,而在各种脑损伤情况下神经细胞外谷氨酸浓度均明显升高,抑制系统XC-的转运作用,进而引发铁死亡。细胞摄取胱氨酸是GSH合成的关键步骤,而GSH的产生和维持对于保护细胞免受氧化应激损害至关重要。
erastin作为铁死亡的诱导剂,在诱导的细胞死亡方面与SAS(一种已知的系统XC-抑制剂)作用方式相似,通过使用放射性物质标记半胱氨酸可观察到erastin和SAS明显抑制细胞对半胱氨酸的摄取。因此,erastin似乎充当系统XC-的直接抑制剂,这一结果表明erastin作用机制与半胱氨酸依赖的GSH合成相关,erastin可以通过抑制细胞摄入胱氨酸进而减少GSH的合成,降低细胞本身的抗氧化应激的能力,诱导细胞死亡。目前使用传统的生化方法和先进的代谢组学分析表明,使用erastin治疗可导致细胞内GSH显著耗尽,而β巯基乙醇(β-mercaptoethanol,β-ME)可逆转erastin和SAS介导的致死作用,且这一过程绕过了系统XC-的跨膜转运作用,β-ME通过与细胞外胱氨酸结合形成二硫化合物,经过不同的转运体将其导入细胞内,通过其他途径增强半胱氨酸的摄取。这些发现进一步证明了estrain或SAS诱导的铁死亡中涉及系统XC-的参与。
2.2 直接抑制GPx4导致铁死亡 铁死亡的特征在于毒性脂质ROS的铁依赖性积累(见图1),特别是当膜多不饱和脂肪酸(polyunsaturated fatty acids,PUFAs)由于脂质过氧化物酶谷胱甘肽过氧化物酶4(glutathione peroxidase 4,Gpx 4)失活而失去控制时,铁死亡就会发生。GPx4被认为是脂质过氧化的抑制蛋白,它是一种将H2O2和有机H2O2分解成水或相应的醇的酶,GSH是其活化过程中必不可少的辅助因子。因此,铁死亡诱导剂erastin和SAS可通过耗尽细胞内GSH从而降低GPx4活性,以此提高细胞质和脂质中ROS水平,最终导致细胞发生萎缩、死亡。
GPxs家族由不同的成员组成,包括GPx1-8,其中GPx4在铁死亡中扮演比其它成员更重要的角色。RAS选择性致死性小分子(RAS-selective lethal 3,RSL3)是一种可以直接与GPx4结合并抑制GPx4活性的脂肪细胞诱导剂,它可导致脂质过氧化物在细胞内积累,随后引发铁死亡。除了erastin,其它铁死亡诱导剂也已经在大量的筛选实验发现,这些诱导剂包括DPI7、DPI10、DPI12、DPI13、DPI17、DPI18、DPI19和RSL3等,与RSL3一样,这些诱导剂均可直接抑制GPX4的酶活性而不会耗尽细胞内GSH。因此,RSL3和功能相关的化合物被归类为“2类”铁蛋白诱导化合物(ferritin-inducing compounds,FINs),以区别于erastin和其他系统XC-抑制剂。
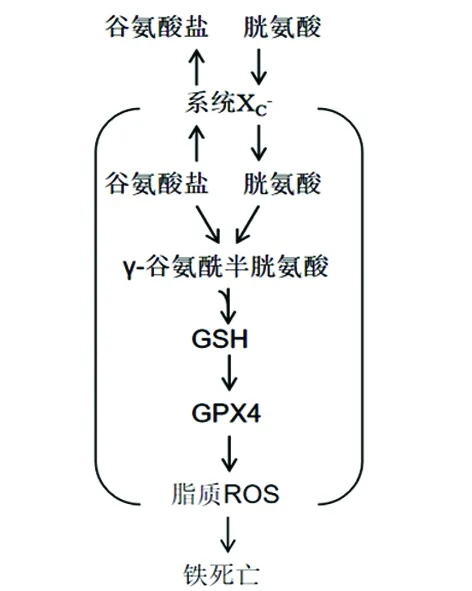
图1 铁死亡作用机制
2.3 其它可能诱导铁死亡的机制 电压依赖性阴离子通道(voltage-dependent anion channels,VDACs),也被称为膜孔蛋白,是真核细胞输送离子和代谢物的跨膜通道,其在线粒体外膜上分布较多。erastin可以与线粒体外膜上的VDAC2和VDAC3结合,改变膜的通透性,减缓NADH的氧化。除此之外,erastin也可改变VDAC的离子选择性,导致通道只允许阳离子进入线粒体内,以引起线粒体功能障碍和氧化剂释放,最终导致氧化依赖性非凋亡性细胞死亡,即铁死亡。
更有研究发现[9,10]p53可通过p53-SLC7A11轴抑制SLC7A11的表达,从而阻碍胱氨酸的摄取,促进细胞凋亡,而p53的作用可以通过铁死亡抑制剂来阻断。此外,p53还增加了细胞内ROS水平并触发ROS诱导的应激反应,最终导致细胞的铁死亡。因此,在癌症的背景下,铁死亡可作为p53通过抑制铁死亡下游的通路发挥抗肿瘤作用。
3 铁在铁死亡中的作用
铁在铁死亡的执行过程中至关重要,膜渗透性铁螯合剂如环吡酮(ciclopirox,CPX),311和2,2-联吡啶(2,2-bipyridyl,2,2-BP)和膜不可渗透铁螯合剂如去铁胺(deferoxamine,DFO)都可有效防止estrain,RSL3以及生理刺激诱导的细胞铁死亡。同样, TFRC是转铁蛋白受体的编码基因,转铁蛋白受体是转铁蛋白-铁复合物摄入细胞所需的物质[11],沉默TFRC编码基因也可有效预防由erastin或胱氨酸缺乏所诱导的铁死亡。反之,补充结合铁的转铁蛋白或可直接利用的铁(如柠檬酸铁铵),但不补充其他二价金属后加速erastin诱导的铁死亡。以上结果明确了铁死亡对铁的需求。尽管目前尚不清楚铁如何促进细胞的死亡,但铁螯合剂可抑制铁死亡,这一结果明显的解释了铁螯合剂阻断铁向氧提供给电子以形成ROS这一过程,进而阻断了铁的氧化还原作用。
铁螯合剂根据其性质不同而发挥不同的作用。亲脂性铁螯合剂可穿过细胞膜并螯合细胞内游离的“氧化还原性”铁池,这通过阻断铁池中催化可溶性或脂质自由基的形成来阻止细胞死亡,这些自由基可以引发或传播氧化性多不饱和脂肪酸碎片。亲脂性铁螯合剂还可直接灭活促进膜脂质氧化的含铁酶。与亲脂性铁螯合剂不同,DFO是一种膜不可渗透的铁螯合剂,其可通过胞吞作用在溶酶体中积累。这表明DFO可能通过螯合溶酶体上的铁来预防细胞铁死亡。然而,与导致溶酶体破坏的致死性H2O2治疗不同,现阶段没有证据可以直接表明铁死亡诱导剂如erastin引发溶酶体死亡。因此,在溶酶体内起作用的DFO可有效截获细胞中其他位置更直接地促进L-ROS形成的铁,以防止细胞死亡。铁螯合剂不仅可以防止铁死亡,还可以防止H2O2和青蒿琥酯引起的细胞死亡。然而,这些致命因素引发的细胞死亡通常不会被特定的特异性抗氧化剂如Fer-1所阻断[12]。因此,在大多数致死性表型中需要联合使用铁螯合剂和亲脂性抗氧化剂来明确评估铁死亡的作用。
4 铁死亡与脑损伤
Dixon等人首次将铁死亡于急性脑损伤联系起来,确立了这两个过程相关的概念。研究发现将解剖的大鼠海马切片暴露于高浓度的谷氨酸下可诱导细胞死亡,而这一过程可通过与铁蛋白抑制素-1或环吡酮的共处理被抑制(约50%)。随后研究发现脑白质损伤与脂质过氧化水平的升高有关且补充铁、抑制GSH的从头合成以及抑制N-乙酰半胱氨酸可加剧细胞死亡,这一过程与铁死亡的机制一致。在少突胶质细胞死亡模型中,ferrostatin-1抑制剂及其改良的类似物可以阻断细胞死亡。此外,谷氨酸培养的少突胶质细胞的致死性受到ferrostatin-1、liproxstatin-1、铁螯合剂和其他已知抑制铁死亡的小分子物质所抑制[13],而Caspase抑制剂Z-VAD-FMK或坏死性凋亡抑制剂necrostatin-1s不会阻断这些条件下的细胞死亡,也不会导致坏死因子复合物的形成,进一步排除细胞死亡过程中凋亡和坏死的参与。
Tuo等人[14]进一步阐明了铁死亡是体内急性病理性脑细胞死亡的机制,在单侧短暂性大脑中动脉闭塞(middle cerebral artery occlusion,MCAO)存在的情况下,tau蛋白、淀粉样前体蛋白(amyloid precursor protein,App)和亚铁转运蛋白(ferrous iron exporter ferroportin,Fpn)与铁水平的升高有关。研究发现在大鼠和小鼠模型中,MCAO可导致损伤半球中铁水平的升高,这种铁的积累可引发铁死亡。而liproxstatin-1或ferrostatin-1(特异性铁死亡抑制剂)治疗可限制梗死面积,减少MCAO引起的行为缺失。MACO通过一种未知的机制引起缺血导致tau蛋白减少;相反,tau蛋白的减少抑制App调节的Fpn依赖性铁输出,而铁输出的丧失导致细胞内铁的积累和对铁死亡的易感性增加。另一方面,脑损伤部位局部铁浓度的升高也可能是有由于损伤区域附近的血管渗漏导致全-转铁蛋白的流入所致[15],增加血液中转铁蛋白的水平(降低转铁蛋白饱和度百分比)可有效减少脑损伤并改善神经功能,这一过程可能与神经元脂质过氧化和细胞死亡减少有关。此外,Zille等人也证明了铁死亡抑制剂在抑制MCAO诱导的脑损伤中的功效[16]。
脑出血后的脑损伤是由血管破裂和血液渗入脑中引起,这一过程也涉及铁死亡。血液中血红蛋白所含的铁可以通过增强ROS形成和释放引起神经元损伤[4]。Ferrostatin-1或liproxstatin-1可以抑制用血红蛋白或游离铁处理的脑切片培养物海马区域中的细胞死亡。暴露于血红蛋白结合的铁导致这些海马细胞中GSH耗尽和GPX4失活。在胶原酶诱导的血管损伤的体内模型中,在损伤部位或远端直接注射铁卟啉菌抑制剂可减少受损细胞的数量和损伤的大小,改善动物的神经功能[4]。细胞内半胱氨酸可以促进GSH的产生,细胞渗透性半胱氨酸类似物N-乙酰半胱氨酸(N-acetyl-cysteine,NAC)可以保护脑组织免受脑出血相关细胞死亡的影响[17];因此,可认为脑内出血后的铁死亡是由GSH合成缺陷引起的。
此外,铁死亡也与创伤性脑损伤有关。在创伤性脑损伤的大鼠皮质-撞击模型中,损伤1 h后可检测到氧化的PE脂质水平升高,与GPX4功能的丧失一致[18]。在创伤性脑损伤的小鼠皮质-撞击模型中,在损伤部位处及其附近可观察到铁阳性细胞的减少[19]。此外,通过ferrostatin-1治疗发现细胞死亡和相关的行为改变得以减少。实际上,研究发现关于脑损伤中细胞的死亡方式与其所承受的压力类型有关[20]。
5 展 望
关于铁死亡,尽管目前我们在理解其诱导机制和信号传导途径方面的研究进展越来越多,但仍存在各种问题,其中明确铁在铁死亡中的决定性作用将有助于我们理解铁死亡的发生和调节机制。尽管各种研究表明铁死亡与各种疾病有关,如神经退行性疾病、肾衰竭以及肿瘤等,但铁死亡在人类疾病中的作用仍然是一个谜。由于细胞对铁死亡的易感性在不同组织之间变化很大;同样,细胞对铁死亡诱导剂(例如索拉非尼)的敏感性在不同个体之间存在差异。此外,研究铁死亡和脑损伤之间关系的动物实验通常使用抑制剂(如铁蛋白)来阻断细胞死亡,但目前研究尚未阐明大脑复杂环境中诱导铁蛋白沉积的特定信号。因此,未来研究的一个基本领域涉及脑细胞中铁转化过程的生化调节。铁死亡也可能在脑细胞中表现出更大的调节复杂性或与其他脑特异性细胞死亡途径相互作用。
然而铁死亡在脑损伤动物模型中的成功干预,目前主要问题将在于铁死亡抑制剂在临床应用领域中的问题,开始新的临床试验来检测铁死亡抑制剂作为脑损伤期间病理细胞死亡的关键调节因子与否也值得进一步讨论。鉴于铁螯合剂和抗氧化的小分子在治疗脑损伤方面取得了不同程度的成功[21],现将已知的抗氧化能力结合不断补充的铁死亡机制的认识,可有助于鉴定或开发特异性阻断体内铁依赖性ROS积累的化合物,针对这一过程的研究有可能产生对脑损伤的新认识和有效治疗的新途径。
