在明串珠菌中构建葡萄糖到甘露醇的转化体系
2019-07-04金红星王星彭钰玮
金红星,王星,彭钰玮
(河北工业大学 化工学院,天津,300130)
甘露醇(mannitol)是一种具有多种功效的六糖醇,被广泛地应用于医药、食品、化工和电子等行业[1]。目前生产甘露醇的4种方法中,海藻提取法产生大量废水、能耗高、污染严重;催化加氢法在高温高压、金属催化和通氢气条件下进行且产物分离困难[2];酶转化法需要昂贵的辅酶;微生物发酵法产甘露醇绿色清洁,是产业转型升级的迫切需要。产甘露醇的细菌、酵母和丝状真菌中,进行异型乳酸发酵的细菌(lactic acid bacteria, LAB)比较出色[1]。明串珠菌(Leuconostoc)、酒球菌(Oenococcus)和乳杆菌(Lactobacillus)等[3]异型乳酸发酵细菌,将果糖底物转化为甘露醇。国内外的很多学者[4-8]研究了产甘露醇的发酵工艺。然而,南京工大的ZHANG等[9]认为,发酵法产甘露醇途径经济效益低的原因是发酵培养基成本高、果糖的渗漏而进入中心代谢、甘露醇产率低。因此,本课题组[10-11]通过多基因敲除、mdh(甘露醇脱氢酶)基因敲入改造了染色体,并考察了对肠膜明串珠菌发酵产甘露醇的影响,以相对廉价的蔗糖作为底物时,产率已经达到90%以上。
为进一步提高底物到目的产物的转化率,在肠膜明串珠菌中拟引入同型乳酸发酵细菌产甘露醇的代谢途径,即葡萄糖→葡萄糖-6-磷酸→果糖-6-磷酸→甘露醇-1-磷酸→甘露醇[1, 12]。明串珠菌的PPK(戊糖磷酸解酮酶)代谢途径存在2种酶的编码基因,即催化葡萄糖→葡萄糖-6-磷酸的葡萄糖激酶和催化葡萄糖-6-磷酸→果糖-6-磷酸的磷酸葡萄糖异构酶[13]。因此,只要引入催化果糖-6-磷酸→甘露醇-1-磷酸的甘露醇-1-磷酸脱氢酶基因(mt1d)和催化甘露醇-1-磷酸→甘露醇的甘露醇-1-磷酸酶基因(m1p),就可以在明串珠菌中构建由葡萄糖转化为甘露醇的体系。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒
肠膜明串珠菌(Leuconostocmesenteroides)CGMCC1.10327、大肠杆菌(E.coli)DH5α、质粒pUC19,均由本实验室保存;肠膜明串珠菌ATCC8293、植物乳杆菌CGMCC1.2437、食淀粉乳杆菌(Lactobacillusamylovorus)CGMCC1.3395,购自中国普通微生物菌种保藏管理中心。
1.1.2 试剂
高保真的DNA聚合酶、T4DNA连接酶:谦泰生物技术有限公司;PCR纯化试剂盒:Axygen公司;限制性内切酶:大连宝生物工程有限公司;氨苄青霉素:Sbase公司。引物由金唯智生物科技有限公司合成(表1),m1p基因编码序列由生工生物工程有限公司合成。
1.1.3 培养基与培养条件
大肠杆菌用LB培养基培养,培养温度为37 ℃;肠膜明串珠菌和植物乳杆菌用MRS培养基培养,培养温度为30 ℃。
1.1.4 仪器与设备
Mastercycler®nexus PCR仪,Eppendorf;Gene Pluser XcellTM电转化仪,Bio-Rad;LC-20AD CTO-20A高效液相色谱仪,Agilent。
1.2 方法
1.2.1mt1d-m1p串联体的合成
以植物乳杆菌染色体DNA为模板,利用1对引物mt1d1/mt1d2 PCR扩增mt1d编码序列;以肠膜明串珠菌ATCC8293染色体DNA为模板,利用1对引物ldhA1/ldhA2 PCR扩增D-ldh基因的表达元件;通过重叠延伸PCR将D-ldh基因表达元件和mt1d编码序列连接成为mt1d基因表达盒。
根据GenBank中登录号为AF032462的柔嫩艾美球虫(Eimeriatenella)的m1p编码序列,按照明串珠菌染色体的密码子偏好性优化了核苷酸序列,并委托公司人工合成了DNA序列。利用2对引物ldhA3/ldhA4、m1p1/m1p2,通过重叠延伸PCR合成了m1p基因表达盒。
利用1对引物mt1de1/mt1de2 PCR扩增获得的mt1d基因表达盒序列插入到pUC19的EcoRI和XbaI位点上,命名为pUC19-mt1d。利用1对引物m1pe1/m1pe2 PCR扩增获得的m1p基因表达盒序列插入到pUC19-mt1d的KpnI位点上,成为mt1d-m1p表达盒串联体。
1.2.2 同源重组载体的构建
利用2对引物dtsq1/dtsq2、dtsh1/dtsh2,通过重叠延伸PCR获得的产物插入到pUC19的EcoRI和HindⅢ位点上,成为中间带有XbaI识别序列的葡聚糖蔗糖酶基因的同源重组载体。
利用2对引物aldhq1/aldhq2、aldhh1/aldhh2,通过重叠延伸PCR获得的产物插入到pUC19的EcoRI和HindⅢ位点上,成为中间带有XbaI识别序列的乙醛脱氢酶基因的同源重组载体。
以食淀粉乳杆菌染色体DNA为模板,利用1对引物amyl/amyr通过PCR获得的产物插入到同源重组载体XbaI位点上,成为中间带有α-淀粉酶基因标记的同源重组载体。
以mt1d-m1p表达盒串联体为模板,利用1对引物mtmpl/ mtmpr通过PCR获得的产物插入到同源重组载体的XbaI位点上,成为中间带有mt1d-m1p表达盒串联体的同源重组载体。
1.2.3 突变菌株的构建和验证
参照文献[15]的方法进行明串珠菌的电击转化。通过2次同源重组获得mt1d-m1p表达盒串联体定点插入到染色体的突变菌株。第1次同源重组:用中间带有α-淀粉酶基因标记的同源重组载体转化初始菌株,在平板上获得蓝色的目的菌株,即靶基因失活、带有标记基因的菌株。第2次同源重组:用中间带有mt1d-m1p表达盒串联体的同源重组载体转化第1次同源重组获得的目的菌株,在平板上获得白色的目的菌株,即靶基因失活、定点插入mt1d-m1p表达盒串联体的菌株。

表1 本研究中使用的引物Table 1 Primers used in this study
以染色体DNA为模板,利用1对引物aldhyq/aldhyh进行PCR,通过琼脂糖凝胶电泳验证aldh基因失活、带有标记基因的菌株(Δaldh::amy)和aldh基因失活、定点插入mt1d-m1p表达盒串联体的菌株[Δaldh::(mt1d-m1p)]。
1.2.4 发酵产甘露醇
野生型菌株和突变菌株用发酵培养基[14-15]进行摇瓶发酵20 h,用HPLC测定甘露醇含量。参照文献[16-17]改进的检测条件:检测器为蒸发光散射检测器(ELSD 6000),色谱柱为Ultimate XB-NH2,流动相为V(乙腈) ∶V(水)=85∶15,流动相流速为:1 mL/min,柱温为40 ℃,漂移管温度为95 ℃,气流流速为3L/min。
2 结果与分析
2.1 mt1d-m1p串联体的合成
第1轮PCR扩增得到了预期长度为177 bp的D-ldh基因表达元件和1 164 bp的mt1d编码序列(图1-A,图1-B)。第2轮PCR通过2个第1轮PCR产物末端的反向互补序列相互退火结合,构建成D-ldh基因表达元件和mt1d编码序列连接在一起的DNA融合体。但是此时该融合体的量比较少,经过第3轮PCR特异性地扩增,成功得到了预期为1 341 bp的mt1d基因表达盒(图1-C)。

M-Marker;1和2-D-ldh基因表达元件;3-mt1d编码序列;4和5-mt1d表达盒。图1 合成mt1d表达盒的重叠延伸PCRFig.1 Overlapping extension PCR for the synthesis of mt1d expression cassette
第1轮PCR扩增得到了预期长度为177 bp的D-ldh基因表达元件和946 bp的m1p编码序列(图2-A,图2-B)。

M-Marker;1和2-D-ldh基因表达元件;3和4-m1p编码序列;5-m1p表达盒图2 合成m1p表达盒的重叠延伸PCRFig.2 Overlapping extension PCR for the synthesis ofm1p expression cassette
第2轮PCR利用2个第1轮PCR产物末端的反向互补序列相互退火结合,并通过8轮循环使2个DNA片段重叠延伸,经过第3轮PCR特异性地扩增,成功得到了预期为1 107 bp的m1p基因表达盒(图2-C)。
将mt1d表达盒和m1p表达盒的串联体连接到pUC19上命名为pUC19-mt1d-m1p,经EcoRI和XbaI双酶切验证结果(图3)显示,得到了预期为2 659 bp的线性pUC19和2 483 bp的插入片段。pUC19-mt1d-m1p的测序结果表明,插入片段序列与预期结果一致。

M-Marker;1-酶切结果图3 pUC19-mt1d-m1p的酶切验证Fig.3 Digestion verification of pUC19-mt1d-m1p
2.2 同源重组载体的构建
通过PCR验证肠膜明串珠菌突变菌株的琼脂糖凝胶电泳结果见图4,由图4可知,2种突变菌株符合预期,即野生型肠膜明串珠菌菌株扩增的片段大小为1 125 bp、Δaldh::amy菌株扩增的片段大小为3 079 bp、Δaldh::(mt1d-m1p)菌株扩增的片段大小为3 417 bp, 说明在肠膜明串珠菌中成功构建了葡萄糖到甘露醇的体系。
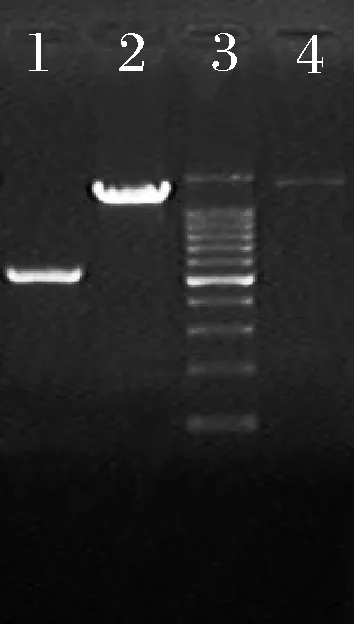
1-野生型菌株(1 125 bp);2-Δaldh::amy(3 079 bp);3-Marker;4-Δaldh::(mt1d-m1p) (3417 bp)图4 PCR验证突变菌株的琼脂糖凝胶电泳图Fig.4 Agarose gel electrophoresis map of mutant strain’s PCR verification
2.3 甘露醇的产量比较
肠膜明串珠菌不同菌株发酵产甘露醇的产量见图5。
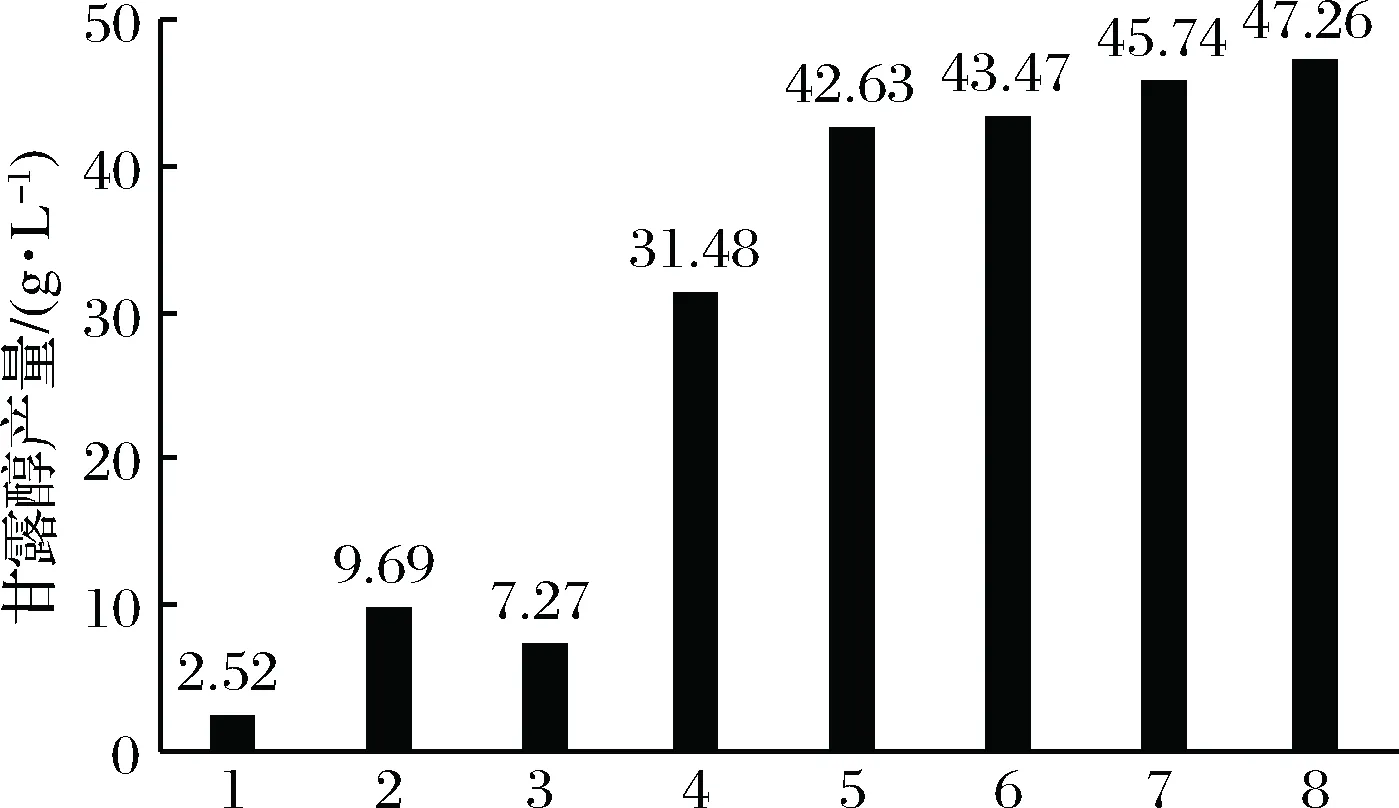
1-20 g/L葡萄糖为底物的Δaldh::(mt1d-m1p);2-20 g/L蔗糖为底物的Δaldh::(mt1d-m1p);3-20 g/L蔗糖为底物的野生型菌株;4-90 g/L蔗糖为底物的野生型菌株;5-90 g/L蔗糖为底物的Δaldh::(mt1d-m1p);6-90 g/L蔗糖为底物的Δaldh::(mt1d-m1p)Δdts::amy;7-90 g/L蔗糖为底物的Δaldh::(mt1d-m1p)Δdts::(mt1d-m1p);8-90 g/L蔗糖为底物的Δdts1ΔD-ldhΔpat::mdhΔstpk::mdh Δfk::mdhΔaldh::(mt1d-m1p)图5 野生型和突变型菌株的甘露醇产量Fig.5 Mannitol yield of wild-type and mutant-type strains
从图5可以看出,(1) 以20 g/L葡萄糖为底物时,Δaldh::(mt1d-m1p)的甘露醇产量为2.52 g/L,异型乳酸发酵细菌-明串珠菌中已经构建成功了同型乳酸发酵细菌产甘露醇的代谢途径;(2) 以20 g/L蔗糖为底物时,Δaldh::(mt1d-m1p)(9.69 g/L)的底物到甘露醇的转化率比野生型(7.27 g/L)提高了33.29%;(3) 以90 g/L蔗糖为底物时,定点插入mt1d-m1p表达盒的菌株转化率比野生型提高了很多,多基因敲除菌株[Δdts1ΔD-ldhΔpat::mdhΔstpk::mdhΔfk::mdhΔaldh::(mt1d-m1p)]产量最高;(4) 以 90 g/L蔗糖为底物时,双拷贝插入mt1d-m1p表达盒的菌株[Δaldh::(mt1d-m1p)Δdts::(mt1d-m1p)]比单拷贝插入菌株[Δaldh::(mt1d-m1p)、Δaldh::(mt1d-m1p)Δdts::amy]产量高,Δaldh::(mt1d-m1p)Δdts::amy比Δaldh::(mt1d-m1p)产量高,这是dts基因(葡聚糖蔗糖酶)失活造成的。
3 讨论
近年来,本课题组一直从事着产甘露醇明串珠菌的遗传育种工作。为了降低生产成本,以蔗糖代替葡萄糖、果糖作为底物进行了发酵产甘露醇的研究[18]。以蔗糖作为底物时,既可以在肠膜明串珠菌胞外由葡聚糖蔗糖酶催化生成葡聚糖和果糖,又可以进入胞内进行代谢[10]。为了使更多的蔗糖进入胞内进行代谢,删除了单拷贝的葡聚糖蔗糖酶基因(dts),从而提高了甘露醇的产量[15, 19]。由甘露醇脱氢酶催化果糖转化为甘露醇时,需要NAD(P)H[10]。因此,删除dts(Δdts)的基础上,进一步删除了代谢过程中消耗NAD(P)H的D-乳酸脱氢酶(D-ldh)和乙醛脱氢酶(aldh)编码基因,进而又提高了甘露醇的产量[20]。肠膜明串珠菌中存在多拷贝的葡聚糖蔗糖酶基因[13],故删除了诱导表达葡聚糖蔗糖酶基因的双组分系统成员-丝氨酸/苏氨酸蛋白激酶[21]编码基因(stpk)[10-11]。肠膜明串珠菌中除了存在乙醛脱氢酶基因外,还有乙醛脱氢酶-乙醇脱氢酶双功能酶编码基因[13],故阻断了PPK代谢途径中乙酰磷酸到乙酰CoA 的反应(催化此反应的酶编码基因为pat)[10-11]。使果糖激酶基因(fk)失活,以便几乎所有的果糖转化为甘露醇[10-11]。将3个拷贝的甘露醇脱氢酶基因(mdh)敲入到肠膜明串珠菌染色体中,以便提高甘露醇脱氢酶的酶活,进而提高产量[10-11]。最终,构建了高产甘露醇的菌株肠膜明串珠菌Δdts1ΔD-ldhΔpat::mdhΔstpk::mdhΔfk::mdh。为了进一步提高蔗糖到甘露醇的转化率,本研究在Δdts1ΔD-ldhΔpat::mdhΔstpk::mdhΔfk::mdh(甘露醇产量为41.0 g/L)基础上,在染色体的aldh位点上定点插入了mt1d-m1p表达盒串联体,从而获得了突变菌株Δdts1ΔD-ldhΔpat::mdhΔstpk::mdhΔfk::mdhΔaldh::(mt1d-m1p)(47.26 g/L),转化率又提高了7%。在前期工作中发现,蔗糖底物为90 g/L时甘露醇产量最高[22],故本研究采用了90 g/L的蔗糖底物。印度的RESHAMWALA等[12]以质粒形式在大肠杆菌过表达了mt1d、m1p和pxtD,因此进行发酵时必须加入抗生素而保持质粒的稳定性。本研究是在染色体上定点插入(mt1d-m1p)的,发酵时不需要加入抗生素。
开始启动本研究时,企图通过重叠延伸PCR将mt1d表达盒和m1p表达盒串联在一起,但是所有的努力都以失败而告终。这是2个表达盒所采用的相同的D-ldh基因表达元件在重叠延伸过程中相互干扰而造成的。通过PCR扩增第一个基因表达盒片段时,在引物中引入酶切位点识别序列,将第二个基因表达盒片段插入到这一位点,从而将2个表达盒串联在一起了。
本研究通过构建葡萄糖到甘露醇的转化体系,使出发菌株大幅度提高了甘露醇产量,但是改造的菌种还没有达到生产中能够采用的水平。微生物发酵生产甘露醇的方法,能否实际应用的瓶颈是生产成本,故从底物和菌株的遗传育种角度还需要深入研究。例如,以甘蔗汁作为培养基[22]或以菊粉[23-25]作为底物,都可以降低发酵成本。
4 结论
以90g/L蔗糖为底物时,mt1d-m1p表达盒串联体的定点插入使初始菌株的蔗糖到甘露醇的转化率提高很多。多基因敲除菌株中定点插入mt1d-m1p表达盒串联体又提高产量。双拷贝插入mt1d-m1p表达盒的菌株比单拷贝插入菌株提高产量。总之,增加甘露醇的合成途径是增产的手段之一。
