芯片液相色谱-质谱法高灵敏度检测水产品中微囊藻毒素
2019-06-21卢巧梅
卢巧梅
(福州大学 测试中心,福州大学食品安全与生物分析教育部重点实验室,福建 福州 350116)
蓝藻水华是常见的淡水水华现象,产生的毒素以微囊藻毒素(MCs)最普遍且危害最大[1-2].大量藻毒素的存在不仅影响生态环境,还会通过食物链富集和传递,影响淡水养殖业并威胁人类健康.MCs是一类由7个氨基酸残基组成的环状多肽化合物.目前已发现了几十种该类毒素,其中MC-RR、MC-LR和MC-YR最具有代表性,其测定技术包括生物学法、免疫检测法和化学分析法等[3].化学分析法中,HPLC是推荐的MCs测定方法.痕量毒素通常需要先浓缩富集再经 HPLC-UV法直接测定[4-5].此法结合质谱检测器,方法的灵敏度和定性准确性明显提高,但较大的进样量和较低的柱效在常规HPLC-MS或HPLC-MS/MS中略显不足[6-7].因此,开发一种高效、样品消耗少的MCs检测方法仍具有实际意义.
芯片色谱技术把传统色谱柱浓缩成约数厘米长、数毫米厚的芯片,再通过激光镭射技术在色谱芯片上刻出分析通道,并填充C18、C8等固定相.该微型化装置可明显降低流速、减少柱体积,从而提高方法灵敏度[8-9].目前芯片液相色谱-质谱(nano-flow chip liquid chromatography-high resolution mass,nano-flow chip LC-MS)技术已成功应用于蛋白质、肽类等生物样品分析[10-12],但较少用于小分子化合物领域[13-15],在MCs定量监测中也鲜见报道.
本文将芯片液相色谱和高分辨质谱技术相结合,建立了同时检测 MC-RR、MC-LR和MC-YR的nano-flow chip LC-MS新体系.方法快速可靠,仅pg级的进样量也获得了较高的灵敏度.
1 试验部分
1.1 仪器与试剂
Nano-flow chip LC-MS联用系统(Agilent,USA)包括纳流液相色谱(1200 Nano HPLC)、接口(Chip Cube)和四级杆-飞行时间质谱仪(G6520 Q-TOF MS)3部分.芯片为Agilent G4240-65005,包括富集柱(体积40 nL)和分析柱(75 μm×43 mm),二者填料均为Zorbax SB-C18,5 μm(如图1所示).

图1 MCs的化学结构(a)和芯片示意图(b)Fig.1 Chemical structure of MCs (a)and schematic diagram of chip (b)
标准品 MC-RR、MC-LR和MC-YR(北京伊普瑞斯公司,结构式如图1所示)使用甲醇∶水(体积比为2∶8)溶解,配制成 100.0 μg/mL储备液,试验前用二次水稀释至所需浓度.乙腈为色谱纯(美国 Sigma-aldrich 公司);醋酸铵为分析纯(天津市福晨化学试剂厂);甲酸(FA)为色谱纯(上海迪马科技有限公司).Bond Elut-C18固相萃取柱(Agilent,USA).试验用水为二次蒸馏水,取自 Milli-Q 超纯水系统.所有溶液使用前均用0.22 μm滤膜过滤.
1.2 芯片液相色谱-质谱条件
毛细管泵流速2 μL/min,泵冲洗选择乙腈-水溶液,10%乙腈等度洗脱.纳流泵流速600 nL/min,乙腈(B相)-水溶液(A相,含0.15% FA).0~20 min,10%~60% B相梯度洗脱.进样体积l μL,进样冲洗体积为 4 μL.
质谱选择正离子模式;m/z 400-1200,干燥气流量3 L/min,雾化气温度 315 ℃,Fragmentor 175 V,毛细管电压调节在1 750 V使喷雾良好.3种 MCs 均测得[M+H]+、[M+2H]2+信号,试验以[M+2H]2+作为定量离子(如表1所列).
1.3 样品处理
明虾和草鱼购于福州当地超市.采用固相萃取法(SPE)提取净化样品,过程在文献[7]基础上稍加改进.准确称取鱼肉或虾肉组织1.0 g,加入5.0 mL 5% 醋酸铵溶液,匀浆搅碎,超声20 min,浸提4 h.过滤,残渣用2.5 mL 5%醋酸铵溶液重复提取两次,合并滤液.分别用甲醇、水活化处理C18小柱后,让滤液以一定速度上样富集藻毒素.待样品全部吸附,以5.0 mL 10%甲醇-水溶液冲洗C18小柱,5.0 mL甲醇洗脱目标物,洗脱液氮吹近干,用二次水定容至1.0 mL,15 000 r/min离心10 min,取上清液进行色谱、质谱分析.

表1 3种 MCs 的保留时间和特征离子Table 1 Retention time and characteristic ions of three MCs
2 结果与讨论
2.1 芯片液相色谱分离优化
芯片液相色谱高度集成了芯片色谱柱分离和预富集、纳流喷雾电离等技术,高度智能化、微型化的同时也具有较好的色谱分离性能.
2.1.1 样品溶剂和上样体积的选择
在芯片中,样品先预富集在富集柱上(柱体积仅40 nL),达到设定的上样体积时,六通阀切换使样品在纳流泵的冲洗下进入分析柱(图1b).若稀释样品的溶剂洗脱强度较大,则可能导致在预富集柱中的样品部分排入废液或后续分离时峰展宽.试验考察了5%乙腈-水、2%乙腈-水、水作为样品溶剂时,样品的分离效果.结果发现,即使添加少量乙腈,3种分析物峰形都略微展宽,最终选择使用水作为样品溶剂.
选择合适的上样体积,对样品的测定非常重要.体积太小,不能保证将所有目标物运送至富集柱;若体积太大,可能会导致弱保留化合物穿透富集柱.鉴于上样体积一般为针座毛细管体积、自动进样器到Chip Cube之间毛细管体积、在线过滤器体积之和的2~6倍,试验选择4 μL为上样体积.
2.1.2 流动相选择
对于芯片色谱而言,流动相对色谱分离的影响甚大.芯片的耐受压力约为15 MPa,若使用甲醇-水体系,较高的压力可能超过芯片的承受范围.使用乙腈-水体系,系统压力更低,而且乙腈具有较强的洗脱能力,能够获得更好的峰形.因此,选择乙腈-水作为流动相.
MCs结构中含有多种氨基酸,其极性会随pH值改变而变化.试验分别考察了甲酸体积分数为0.05%~0.15%时,目标物的分离情况(如图2所示).

图2 不同流动相对分离的影响Fig.2 Effects of different mobile phase on separation(a)乙腈-H2O,(b)乙腈-0.05% FA,(c)乙腈-0.10% FA,(d)乙腈-0.15% FA毛细管泵流速2 μL/min,10%乙腈等度洗脱;纳流泵流速600 nL/min,0~20 min,10-60% B相梯度洗脱进样体积l μL,进样冲洗体积为 4 μL(1)MC-RR(0.5 μg/mL),(2)MC-YR(2.0 μg/mL),(3)MC-LR(1.0 μg/mL)
由图2可见,仅使用乙腈-水二元体系时,MC-YR和MC-LR 基本重合.随着甲酸体积浓度增大,3种物质的分离度增大,响应增强.这是因为随着甲酸浓度的增加,促进了MCs的质子化过程和极性差异情况,从而明显改善了各组分在芯片上的保留行为.因此,最终选取体积分数为0.15%的甲酸作为水相添加剂.
梯度洗脱有助于进一步改善分离效果.尝试了多种梯度洗脱程序(如图3所示),发现增大泵流速或有机相比例,可使保留时间缩短.以20%乙腈为初始有机相冲洗色谱柱时,尽管3种待测物在14 min内完成分离,但柱效和灵敏度都明显下降.兼顾分离效率和方法灵敏度,选择0~20 min、10%~60% B作为后续分析条件.

图3 不同梯度洗脱条件对分离的影响Fig.3 Effects of different gradient elutions on separation乙腈-0.15% FA;毛细管泵流速2 μL/min,10%乙腈等度洗脱;进样体积1 μL,进样冲洗体积为 4 μL(1)MC-RR(0.5 μg/mL),(2)MC-YR(2.0 μg/mL),(3)MC-LR(1.0 μg/mL)
2.2 质谱检测
在芯片液相色谱-质谱中,流动相组成、芯片喷针位置、芯片差异等因素都会影响纳流喷雾效果.在色谱分离方面,已证实流动相中添加少量FA有利于改善喷雾情况.试验继续考察芯片因素对分离检测的影响,结果显示,微调芯片喷针位置,目标物的响应信号会随之改变.故所有试验应确保在同一个芯片上进行,且喷针位置一经确定后不再改变.根据喷雾状态优化毛细管电压(1 500~2 000 V),最终确定为1 750 V,在此条件下方法灵敏度更高.
MCs结构中含有氨基、羧基和酰胺基,理论上对质谱的正、负两种离子检测模式都有响应.试验优化了上述两种检测模式下的物质信号情况,也证实了含氮杂原子的存在,使得正离子检测模式下目标物的响应峰面积更高.3种 MCs均测得[M+H]+、[M+2H]2+信号,选择响应较强的[M+2H]2+作为定量离子(如图4所示).在高分辨质谱中,所有目标物的质量偏差更小,定性准确性更强.

图4 正离子检测模式下3种 MCs的全扫描质谱图Fig.4 Mass spectra of three MCs under full scan with positive mode
2.3 定量方法建立
配制一系列不同浓度的MCs标准混合溶液.选择 [M+2H]2+的峰面积定量分析,绘制标准工作曲线.3种MCs在2.5~2 000 ng/mL范围内线性良好,相关系数大于 0.996 5,检测限(LOD)最低为 0.5 ng/mL(如表2所列).目前,我国对水产品中MCs残留未有明确限量,但已规定在饮用水中MC-LR限量值为1.0 ng/mL[16].该方法MC-LR的检测限为0.8 ng/mL,因此能满足痕量MCs日常监测的要求.
考察方法的重现性.日间精密度以当天7 次重复进样,日间精密度以连续3天进样.结果表明,MCs的保留时间稳定,峰面积RSDs小于 6.81%,方法重现性良好.
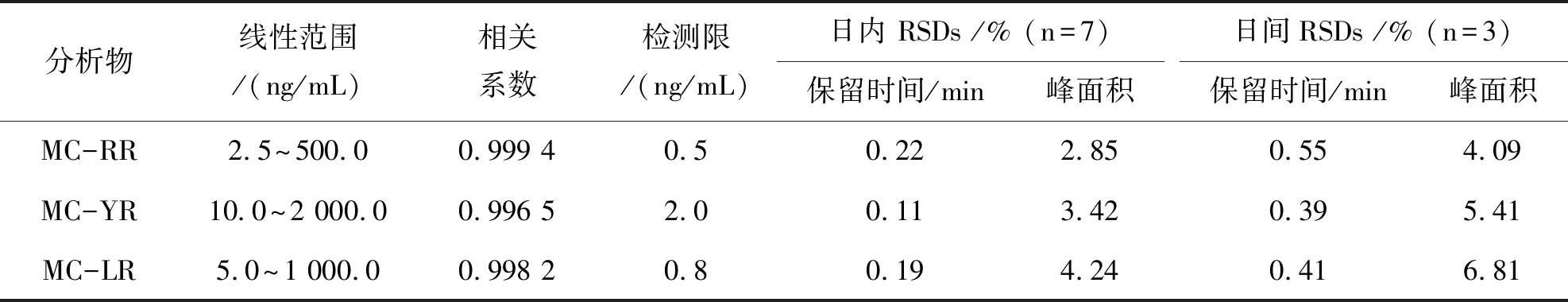
表2 3种 MCs的分析参数Table 2 Analytical parameters of three MCs
2.4 水产品样品分析
分别对鱼肉和虾肉按照1.3节方法进行处理.样品组织经醋酸铵提取、C18净化富集进行质谱分析.对照目标物保留时间、精确质荷比等信息,表明在实际样品中未检出MCs.在最优条件下,进行加标回收率试验(如表3所列).所有样品的回收率大于82.8%~120.3%,能够满足分析方法的要求.

表3 水产品的加标回收率Table 3 Spiked recoveries of aquatic products
2.5 方法对比
将该方法与已报道的HPLC有关方法相比较(如表4所列).由表4可见,在进样体积方面,本法的进样体积远低于其他方法,这对于某些昂贵或稀少样品特别重要,并符合绿色化学的要求.其次,结合经典的SPE预处理方法,本试验的灵敏度可达pg级,线性范围更广.常规的HPLC-UV法通常需要结合一些新兴样品前处理技术(例如固相微萃取和磁性固相萃取)和制备新型吸附材料,才能获得和本方法接近的灵敏度.此外,串联质谱技术具有选择离子、提取离子等功能,可适用于复杂样品分析.综上所述,本法集成样品预富集、分离、喷雾于一块芯片上,操作简单、快速,并具有样品消耗量少、检测限低等突出优势.

表4 本法与HPLC相关方法比较Table 4 Comparison of related HPLC methods
3 结论
试验利用nano-flow chip LC-MS 技术,建立了同时分析3种微囊藻毒素的方法.方法检测限在0.5~2.0 ng/mL范围内.采用固相萃取法纯化2种水产品,均没有测得藻毒素残留.方法简单、灵敏、环境友好,可推广至其他复杂基质中MCs的快速准确分析.
