自修复微胶囊的制备及其影响因素研究
2019-06-19刘兰轩曹东萍刘秀生
郭 靖 ,高 翔,刘兰轩,曹东萍,汪 洋 ,刘秀生 ,季 创
(1.武汉材料保护研究所有限公司,湖北武汉 430030;2.特种表面保护材料及应用技术国家重点实验室,湖北武汉 430030;3.北京星航机电装备有限公司,北京 100074)
0 引言
自修复涂层本质上是能修复如纳米级或微小划痕和裂纹损伤的智能材料,能提高涂层使用寿命和性能。自修复涂层的自我修复属性有内在和外在机制。内在自我修复,指聚合物基质本身包含潜在的自修复功能,如热可逆反应、氢键、离子排列及分子扩散和纠缠触发修复[1-2];外在自我修复,是通过不同的方式,如空心纤维[3-4]、毛细血管[5-7]或微胶囊[8-11]将愈合剂材料引入或预先嵌入聚合物基质中,愈合剂从壳体中释放出来通过不同的修复机制修复裂纹。使用含有愈合剂的微胶囊是设计自愈合涂层的普遍方法。White等[12]在2001年最先提出微胶囊技术,将愈合剂包覆于高分子材料内,形成具有核壳结构的复合材料。在过去十年中,已经开发出含有微胶囊的防腐涂层,用于保护基材免受腐蚀环境(氧气,水,酸和气体)的影响[13-14]。另外,关于自修复脲醛微胶囊的研究也不少,Li等[11]以环氧树脂为芯材、脲醛树脂为壁材,采用原位聚合法制备微胶囊,研究了微胶囊的表面形貌、化学结构、粒径分布及热性能。Blaiszik等[15]以环氧树脂为芯材,氯苯、苯乙酸、苯乙酸乙酯为稀释剂,脲醛树脂为壁材,通过原位聚合法制备微胶囊,研究了不同稀释剂种类及用量下制备的微胶囊的性能。徐梦漪等[16]采用不同相对分子质量的HSMA(苯乙烯马来酸酐共聚物)为乳化剂,通过原位聚合法制备了以石蜡为芯材、三聚氰胺-甲醛树脂(MF树脂)为壁材的相变微胶囊。研究表明,使用高相对分子质量的HSMA为乳化剂制得的微胶囊成球性好,表面光滑,粒径小且分布较窄;使用低相对分子质量的HSMA为乳化剂制得的微胶囊表面粗糙,粒径大且分布较宽;随着HSMA相对分子质量的增加,制得的微胶囊的热稳定性提高。
目前关于解决脲醛微胶囊黏连问题的研究较少,本研究通过改变壁材与芯材的比例等工艺条件来解决脲醛微胶囊的黏连问题。通过原位聚合法制备以环氧树脂(E-51)和环氧小分子(SM80)为芯材,脲醛树脂为壁材的微胶囊,对不同乳化剂条件下制备的微胶囊的微观形态进行分析,考察了不同的乳化速度对微胶囊粒径分布的影响。并讨论了芯/壁比对微胶囊分散情况的影响。
1 试验部分
1.1 试验药品
环氧树脂(E-51),岳阳巴陵华兴石化有限公司;环氧小分子(SM80),江苏三木化工股份有限公司;甲醛(质量分数为37%),分析纯,天津市大茂化学试剂厂;三乙醇胺、无水乙醇,分析纯,天津市天力化学试剂有限公司;阿拉伯胶(GA),上海山浦化工有限公司;尿素、十二烷基苯磺酸钠(SDBS),分析纯,天津市凯通化学试剂有限公司;十二烷基磺酸钠(SDS),分析纯,天津市津北精细化工有限公司;聚氧乙烯脱水山梨醇单油酸酯(吐温-80),化学纯,国药集团化学试剂有限公司;盐酸,分析纯,开封东大化工有限公司试剂厂;蒸馏水,自制。
1.2 微胶囊的制备
将5.35 g尿素和12.57 g甲醛加到250 mL三口烧瓶中,待尿素完全溶解后,加3滴三乙醇胺调节pH至8.5,并加热至65℃,缓慢搅拌下保温反应1 h,得到黏稠液体,加入相对于预聚体2倍质量的蒸馏水,形成稳定透明的脲-甲醛预聚体,备用。
称取一定量的乳化剂、2 g环氧树脂(E-51)与环氧小分子稀释剂的混合液(质量比为0.1),以及蒸馏水(与环氧树脂及环氧小分子稀释剂质量比为20)加入到烧杯中,在一定转速下,乳化30 min,得到芯材乳液。将乳化后的芯材按设计好的壁厚的量倒入250 mL的三口烧瓶中,在转速500 r/min下,将预聚体缓慢滴加到芯材中,用0.1 mol/L盐酸调节pH至2~3,于30 ℃反应0.5 h后,升温至60 ℃,恒温反应1.5 h。将所得产物用抽滤的方式,分别用去离子水、乙醇反复洗涤3遍,将得到的微胶囊放在烘箱里于40 ℃烘4 h,称重,再于60 ℃烘2 h,称重。直至测得的质量差值≤0.000 3 g为止。
1.3 性能检测及表征
(1) SEM表征:将制得的微胶囊样品用双面胶粘附于样品盘上,并在样品表面喷金,采用SU8010型场发射扫描电子显微镜观察微胶囊的表面形貌和分散状态。
(2) 采用DMM-400C型光学显微镜(上海蔡康光学化学有限公司)观察微胶囊乳液的形态和分散状态。
(3) 采用Nicolet iS10型傅里叶变换红外光谱仪对微胶囊进行官能团结构分析。将微胶囊与溴化钾(KBr)混合,研磨成粉末后压片测量。
(4) 粒径测试:采用英国马尔文Nano ZS90型激光粒度分布测试仪对微胶囊的粒径分布及平均粒径进行分析。
2 结果与讨论
2.1 微胶囊的形成过程
尿素和甲醛的合成反应是一个复杂的过程,一般情况下可分为2个阶段:一是UF(脲-甲醛)预聚体的合成;二是羟甲基脲缩聚反应生成脲醛树脂。UF预聚体对微胶囊的形成有重要影响。尿素与甲醛在65 ℃,pH 8.5的弱碱性条件下发生加成反应,形成水溶性的一羟甲脲和二羟甲脲预聚体。UF预聚体中存在大量游离的羟甲基、氨基及亚氨基等活性基团,调节pH,在酸的催化下,主链上剩余的羟甲基与其他主链上的氨基或亚氨基反应,或通过羟甲基之间缩水聚合形成线性或支链型的低分子反应物,经加热固化形成交联网状结构的非水溶性脲醛树脂[11]。
在光学显微镜下观察微胶囊的形成过程,主要有3个阶段:(1)pH调节前,微胶囊形貌与乳液形貌一样,如图1a所示;(2)pH为2~3时,包覆在环氧液滴上的脲醛树脂颗粒小,数量少,随着合成时间的增加,微胶囊逐渐形成,囊壁变厚,如图1b、c、d所示;(3)反应进行到2 h时,芯材已经被完全包覆在脲醛树脂中,形成一定厚度的囊壁,结构致密,如图1e所示。

图1 脲醛微胶囊形成过程的光学显微图Figure 1 Optical micrograph of the formation of urea-formaldehyde microcapsules
2.2 乳化剂对微胶囊性能的影响
2.2.1 乳化剂种类对微胶囊表面形貌的影响
本研究分别采用十二烷基磺酸钠(SDS)、阿拉伯胶(GA)、十二烷基苯磺酸钠(SDBS)和吐温-80作为乳化剂,用量为芯材质量的3%,乳化转速为2 000 r/min,用SEM对微胶囊的微观形貌进行分析,结果如图2所示。由图2a可知,使用SDBS作为乳化剂时,微胶囊部分成形,粘结严重,且仍有较多的脲醛树脂自聚成块。这可能是因为SDBS具有较高的表面活性,乳化能力很强,液滴表面完全被表面活性剂分子所占据,分散相油滴处于相当稳定的状态,当脲醛树脂粒子在芯材表面沉积时,无法抵消空间排斥作用,造成界面上力的不平衡,从而破乳以致不能很好地包封芯材形成胶囊,而是粘结在一起;由图2b、c可知,使用阿拉伯胶和SDS作为乳化剂时,形成的微胶囊都极少,脲醛树脂自聚成块;由图2d可以看出,使用吐温-80作为乳化剂时,微胶囊呈明显的球形。
采用原位聚合法制备微胶囊时加入吐温-80,乳液体系O/W稳定性高,有利于活性预聚体R-NHCH2+富集到液滴表面反应形成囊壁,制备的微胶囊表面光滑致密,水相中未形成囊壁的脲醛树脂颗粒较少,得到形状规则的微胶囊。因此,本研究中采用吐温-80作为微胶囊乳液的乳化剂。
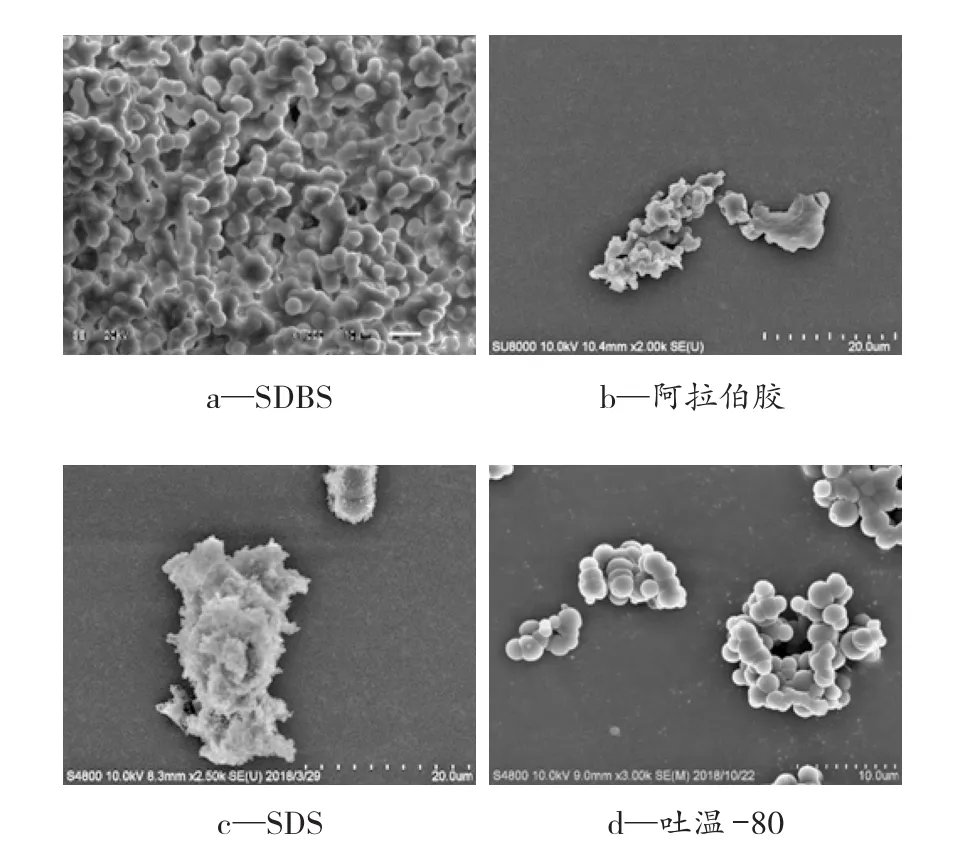
图2 不同乳化剂制备的微胶囊的SEM图Figure 2 SEM photographs of microcapsules prepared with different emulsifiers
2.2.2 乳化剂用量对微胶囊性能的影响?
当乳化剂的用量达到临界胶束浓度时,界面张力不再随乳化剂用量的增大而降低,继续增加乳化剂用量其结构会由球形胶束转变为棒状胶束,因此,乳化剂的用量对微胶囊的性能起着重要作用。在乳化转速为2 000 r/min条件下,采用扫描电子显微镜对吐温-80用量分别为芯材质量的3%、5%和8%时合成的微胶囊的形貌进行分析,结果如图3所示。

图3 吐温-80不同用量下制备的微胶囊的SEM图Figure 3 SEM photographs of microcapsules prepared with different Tween-80 concentrations
从图3中可以看出,当吐温-80用量从3%增加至8%时,芯材都能被包封,用量3%的微胶囊粒径明显大于用量5%和8%的微胶囊,且吐温-80用量大于3%时,微胶囊的粒径变化不大,可能是因为吐温-80的用量过高时,导致乳液黏度过大,不利于胶囊的形成,同时微胶囊团聚严重。因此,本研究中吐温-80的最佳用量为芯材质量的3%。
2.3 乳化转速对微胶囊性能的影响
吐温-80用量为芯材质量的3%,采用激光粒度仪对不同乳化转速下稳定乳液的粒径分布进行分析,结果如图4、5所示。
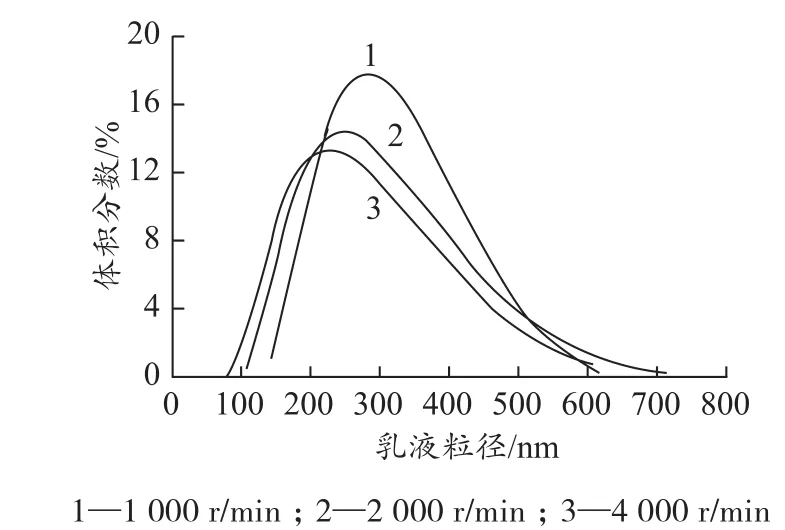
图4 不同乳化转速对乳液粒径分布的影响Figure 4 Effect of different emulsification speeds on the particle size distribution of emulsion
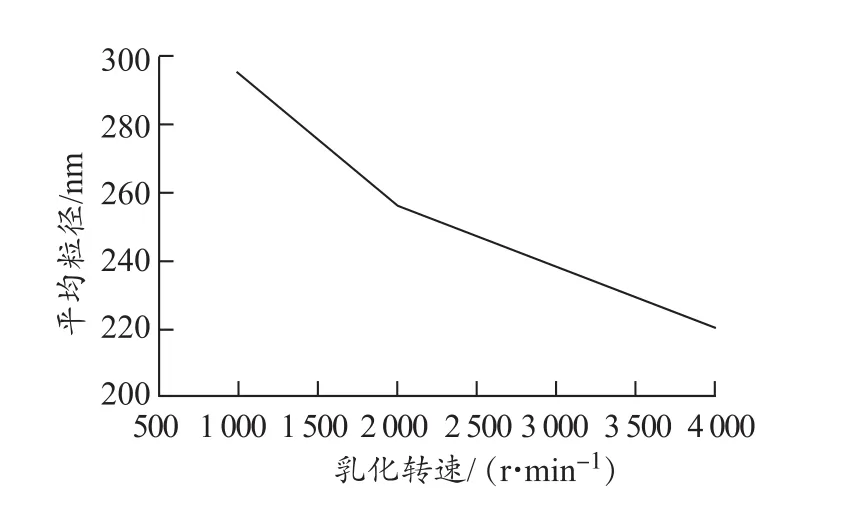
图5 不同乳化转速对乳液平均粒径的影响Figure 5 Effect of different emulsification speeds on the average particle size of the emulsion
由图4、5可以看出,当乳化转速从1 000 r/min增加至2 000 r/min时,微胶囊的平均粒径减小且分布宽度变窄。当乳化转速从2 000 r/min增加至4 000 r/min时,乳液粒径变化趋于平缓。当乳化转速较小时,没有足够的剪切应力将液滴分散,导致乳化不充分,粒径分布宽;当乳化转速增加时,剪切力增大,芯材充分乳化,微胶囊粒径变小且分布变窄;当乳化转速过大时,将会导致乳液破乳,不稳定,且设备动力损耗大,成本高。同时,结合图6中的SEM分析,本研究选择乳化转速为2 000 r/min。
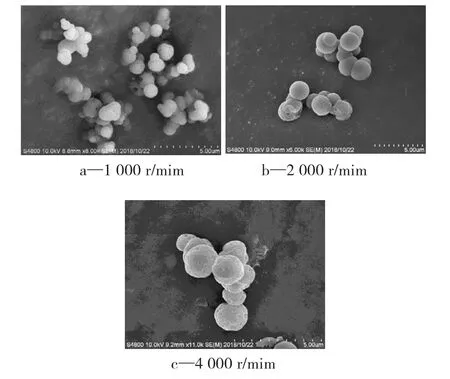
图6 不同乳化转速下微胶囊的SEM图Figure 6 SEM photographs of microcapsules at different emulsification speeds
2.4 微胶囊的化学结构分析
采用傅里叶红外光谱仪分析用吐温-80作为乳化剂时所制备的微胶囊的化学结构,结果如图7所示。图7为干燥的脲醛树脂包封环氧树脂微胶囊产品的红外吸收曲线。从图7中可以看出,3 284 cm-1附近出现了强烈的N—H特征吸收峰,说明体系中存在着丰富的氨基基团;2 965 cm-1处为C—H的特征吸收峰;1 626 cm-1处为碳氧双键羰基特征吸收峰;1 546 cm-1处为C—N的特征吸收峰,这4个吸收峰分别对应了脲醛树脂中的化学键,表明体系中已经合成了脲醛树脂。1 240 cm-1处为环氧基团的对称振动吸收特征峰,1 019 cm-1处为环氧基团非对称吸收特征峰,说明体系中存在环氧树脂,且其结构没有发生变化。
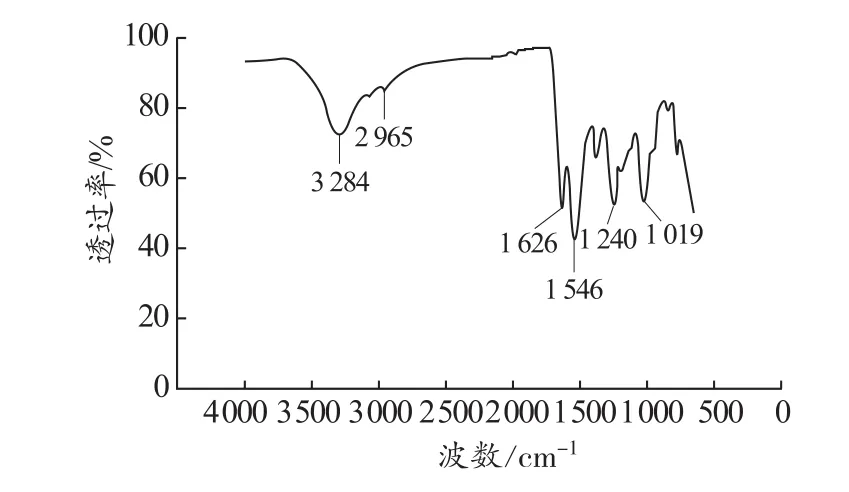
图7 微胶囊的红外光谱图Figure 7 FTIR spectrum of microcapsules
2.5 微胶囊黏连问题分析
制备微胶囊的过程中,当芯材相对过量时,形成的囊壁较薄甚至不能包覆芯材;而当壁材相对过量时,导致囊壁较厚,形成的微胶囊分散性差。所以,选择合适的芯/壁比对微胶囊的制备及其性能非常重要。不同芯/壁比制备的微胶囊的SEM图见图8。

图8 不同芯/壁比制备的微胶囊的SEM图Figure 8 SEM photographs of microcapsules with different core-wall ratio
由图8可知,当芯/壁比为0.2∶1时,形成的微胶囊较少,分散性差,团聚量较多,这可能是因为壁材相对过量,大量的聚脲团聚导致的;当芯/壁比为0.4∶1和0.6∶1时,形成的微胶囊较多,球形规则完整,分散性较好,基本看不到团聚现象;当芯/壁比为1∶1时,微胶囊的分散性变差,团聚量增多,这可能是由于芯材过量,囊壁不能完全包覆芯材。
本研究采用芯/壁比为0.6∶1,此时制备的微胶囊分散性较好,有利于在基体材料中的分布。
3 结语
本研究采用环氧树脂(E-51)和环氧小分子(SM80)为芯材、脲醛树脂为壁材,采用原位聚合法制备了自修复微胶囊。采用光学显微镜考察了微胶囊的形成过程,利用扫描电子显微镜研究了乳化剂种类和用量对微胶囊形貌的影响,同时用激光粒度仪检测了不同乳化转速下制备的微胶囊的粒径分布,得出如下结论:
(1) 在相同工艺条件下,采用吐温-80作为乳化剂,合成的微胶囊球形度较好,外壳光滑,密封良好,脲醛树脂颗粒团聚少。
(2) 随着乳化转速的增加,微胶囊粒径变小,粒径变化趋于平缓;当吐温-80用量为3%,乳化转速为2 000 r/min时,微胶囊粒径分布窄。
(3) 傅里叶红外光谱分析表明,芯材[(E-51)及(SM80)]能良好包覆于脲醛为壁材的微胶囊中。
(4) 通过改变芯/壁比,调整制备脲醛微胶囊的工艺条件,当芯/壁比为0.4∶1、0.6∶1时,得到的微胶囊无黏连,分散性较好。
