测定饲料中维生素A和维生素E含量方法的研究
2019-06-19裘丞军任玉琴
裘丞军, 任玉琴
(浙江省兽药饲料监察所,浙江杭州 311199)
维生素A和维生素E是维持动物体正常代谢所必需的脂溶性维生素,具有促进生长,提高免疫力等作用,已经广泛应用于大规模畜禽养殖中。在饲料添加剂中主要以维生素A乙酸酯和dl-α-生育酚乙酸酯的形式存在,并且通过各种壁材包被保护(杨优敏,2016)。目前测定饲料中维生素A和维生素E的方法已有国家标准GB/T 17812-2008和GB/T 17817-2010,主要是皂化萃取法和甲醇直接提取法,还有碱性蛋白酶水解法(赵晓华等,2010)。皂化萃取法操作步骤繁琐,时间长,消耗试剂多;甲醇直接提取法和酶解法结果不稳定,有些包被产品未能被有效提取。因此本研究拟建立一种能稳定、快速、准确、同时测定饲料中维生素A和维生素E的方法,通过氢氧化钾溶液皂化,乙醇提取,将包被物质溶解,维生素A乙酸酯和dl-α-生育酚乙酸酯转化为维生素A醇和dl-α-生育酚,用外标法计算含量。
1 材料与方法
1.1 试剂和仪器 维生素A乙酸酯标准品(德国Dr.Ehrenstorfer公司);维生素E乙酸酯标准品(Fluka公司);无水乙醇、抗坏血酸、冰乙酸、氢氧化钾均为分析纯;甲醇为色谱纯;水是超纯水;配合饲料、浓缩料、维生素预混料、复合预混料为日常检测时收集。
Agilent1260高效液相色谱仪,紫外可变波长检测器(美国Agilent公司);KQ-500E超声仪(昆山超声仪器公司);ZP205电子天平(Mettler公司)。
1.2 色谱条件 色谱柱:Agilent XDB-C18柱,柱长150 mm,内径 4.6 mm,粒径 5 μm;流动相:甲醇+水=98+2;流速:1.2 mL/min;进样量:20 μL;柱温:30 ℃;紫外检测波长:0~7min,326nm;7~20min,280nm。
1.3 饲料样品前处理 准确称取饲料样品适量(不大于 2 g,精确至0.1 mg),置于 50 mL容量瓶中,依次加入无水乙醇6 mL,10%抗坏血酸水溶液 2.0 mL,470 g/L氢氧化钾水溶液 2.0 mL,混匀,置65℃水浴超声波皂化反应20 min,取出,立即加入1.0 mL冰乙酸调pH至6.0~7.5,冷却至室温,用无水乙醇定容至刻度,混匀,静置。
1.4 标准储备溶液配制 精密称取标准品维生素A乙酸酯62.19 mg,维生素E乙酸酯207.58 mg,分别置于100 mL棕色容量瓶中,加入无水乙醇超声波助溶,冷却至室温,用无水乙醇定容至刻度,混匀,得到维生素A浓度为1807.8 IU/mL,维生素E浓度为2075.8 μg/mL,-20℃避光保存。
1.5 标准工作液配制 精密移取维生素A储备液和维生素E储备液适量,分别置于 50 mL容量瓶中,按照1.3方法处理,用无水乙醇稀释定容成维 生 素 A 浓 度 0.03616、0.1808、0.3616、1.085、5.424、27.12、54.24、108.5 IU/mL 和维生素 E 浓度0.62274、1.2455、6.2274、12.455、24.910、62.274、124.55 μg/mL系列标准工作液。
2 结果与分析
2.1 色谱条件的确立 参考国家标准GB/T 17817-2010和GB/T 17812-2008,维生素A和维生素E的分析方法有正相和反相两种色谱体系。正相法分离效果好,能够有效分离异构体,并且试样中溶解的脂肪容易被流动相冲洗出色谱柱,但是作为溶剂的正己烷挥发性比较大,长时间分析样品浓度会变化,精密度相对较差;反相法稳定性高,对于饲料中维生素A和维生素E的分离效果好,保留时间适中(见图1)。因此选择反相色谱系统,采用 C18 柱,甲醇和水(98∶2)作为流动相,利用紫外检测器可切换波长的功能在326 nm和280 nm处分别记录维生素A和维生素E的色谱峰。

图1 维生素A、维生素E标准图谱(a)和样品溶液图谱(b)
2.2 提取剂的确定 本方法对比选用甲醇、乙醇作为提取剂,实验表明乙醇比甲醇的提取效果好,同时对人体危害性小,因此选择乙醇作为提取剂。
按照本方法,饲料样品前处理过程分为皂化提取和乙醇稀释定容两部分。超声波水浴加热皂化反应时体系中水和乙醇的比例对于一部分饲料中维生素的检出值影响比较大。尤其是水产料中的水分散性维生素颗粒,低含水量皂化液使得饲料黏稠不易分散,维生素A和维生素E检出值相对较低;相反,浓缩料和配合料在水含量高时容易黏稠不易分散。实验比较分析含水量分别为13%、20%、28%、40%、53%五种乙醇皂化体系,结果如表1,当皂化体系水分含量为40%和53%时,提取效果较好。
由于流动相是高比例甲醇体系,如果上机溶液水分含量较高,一些溶于水不溶于甲醇的物质在进样时容易析出,从而污染色谱柱。用水分含量分别为5%、10%、16%三种上机乙醇溶液与甲醇1∶1混合,研究最终定容后上机溶液水分含量对于色谱系统的影响。发现16%溶液产生浑浊,10%和5%溶液澄清。

表1 某水产维生素预混料(水分散型)皂化体系比较
综合分析,确定皂化时水含量为40%,最终定容上机液水分含量为10%,即在50 mL容量瓶中,加入无水乙醇6 mL,抗坏血酸水溶液2.0 mL,氢氧化钾水溶液2.0 mL,然后皂化提取,加入1.0 mL冰乙酸,冷却至室温,用无水乙醇定容至刻度。
2.3 抗氧化剂选择 在高温强碱的环境中维生素E特别容易被氧化,因此在皂化提取时一般加入抗坏血酸(张艳海等,2015;谢云峰等,2014;黄诚等,2012)或者没食子酸(李颂群等,2013)防止维生素氧化,国家标准GB/T 17812-2018和GB/T 17817-2010中还加入了2,6二叔丁基对甲酚(BHT)。本方法采用抗坏血酸作为抗氧化剂,在皂化提取液中分别加入2~200 mg的量,其他条件按照1.3方法处理,比较维生素A和维生素E的值。结果如表2,当抗坏血酸少于40 mg时,维生素E全部或部分损失,维生素A部分损失;当加入量大于100 mg时,维生素A和维生素E的值相对稳定,本方法选取抗坏血酸加入量为200 mg,即2.0 mL10%的抗坏血酸水溶液。

表2 抗坏血酸含量对维生素测定结果的影响
2.4 皂化提取加碱量控制 取5份相同饲料,分别加入1.5~60 mmol不等的氢氧化钾,其他条件按照1.3方法,分析色谱峰。结果如表3,随着碱含量的增加,皂化液pH不断升高,当氢氧化钾低于3.0 mmol时,维生素E未皂化或未完全皂化,维生素A未完全皂化;当氢氧化钾大于10 mmol时,维生素A和维生素E完全皂化,并且值相对稳定。因此,为保证皂化效果,选择加入2.0 mL 470 g/L氢氧化钾(约16 mmol),最后皂化完成时用1.0 mL冰乙酸调pH为6.0~7.5。

表3 不同碱含量对维生素测定结果的影响
2.5 皂化温度控制 取5份相同的饲料,分别在50~100℃的水浴中超声加热,其他条件按照1.3方法,分析色谱峰,计算维生素A和维生素E的含量。结果如表4,维生素A在60~70℃时含量最大,低于60℃或高于70℃含量都偏低;维生素E在50~70℃时含量较稳定,80℃以上时含量下降。分析原因,维生素A在高温下会发生异构和聚合反应(俞安,2012),维生素E在碱性液体高温条件下易氧化降解;同时维生素包膜在温度较低时不易被溶解分散,影响维生素溶出效率,所以根据实验结果选择最佳提取温度为65℃。

表4 皂化温度对维生素测定结果的影响
2.6 皂化时间确定 取A,B两批不同类型饲料样品,每批7份,在65℃水浴中分别超声5~120 min,其他条件按照1.3方法,计算含量,结果如表5,在120 min内提取的维生素含量基本不变,性质较稳定。考虑到其他饲料中维生素含有不同的包被,皂化时间太短不容易完全释出,因此选择20 min作为最终皂化时间。
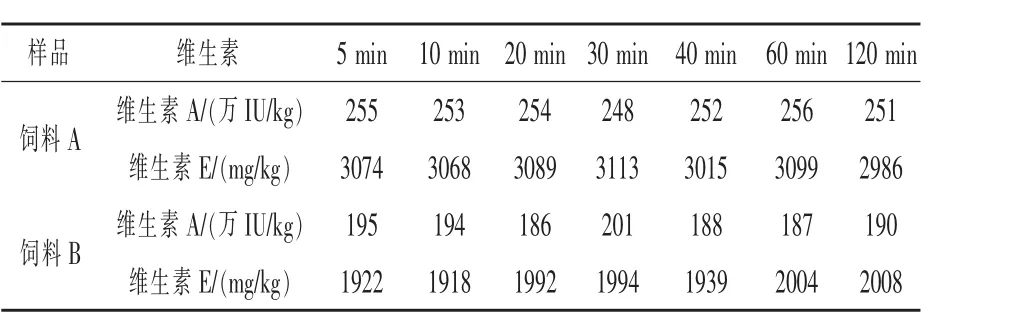
表5 皂化时间对维生素测定结果的影响
2.7 方法线性和检测限 按配制好的系列标准工作液浓度从小到大,分别取20 μL注入液相色谱测定分析,记录峰面积,以浓度(x)为横坐标,峰面积(y)为纵坐标,绘制线性回归曲线;同时添加适量的维生素A和维生素E标准储备液至2 g空白饲料中,按照1.3方法处理,高效液相色谱分析并记录色谱峰,以信噪比大于10确定定量限,结果见表6。

表6 维生素A和维生素E的标准曲线
2.8 回收率和精密度 取三批不同的空白饲料样品,每批5份,每份2 g,分别添加适量的维生素A和维生素E标准储备液,制得低、中、高三种浓度的样品,按实验方法1.3处理,取20 μL样品溶液注入液相色谱测定分析,外标法计算含量,并计算回收率,结果见表7。
2.9 稳定性与重复性 取一份饲料样品按照1.3方法处理,放置 0、1.5、3、4.5、10 h 后进样测定,记录维生素A和维生素E的峰面积并计算RSD分别为0.8%和1.1%,表明饲料样品溶液中维生素A和维生素E在10 h内稳定。另取一批次饲料样品6份,按照1.3方法处理,分别进样测定,计算维生素A和维生素E含量,计算RSD分别为2.5%、4.9%,表明本方法重复性良好。
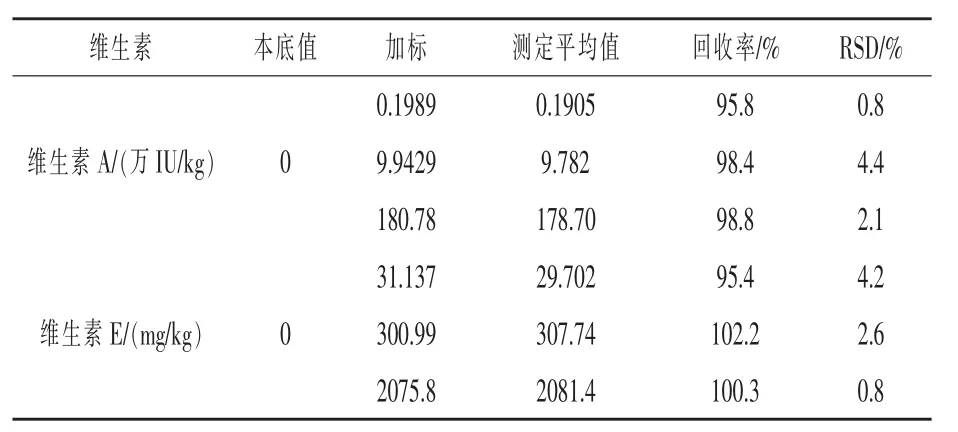
表7 回收率实验结果(n=5)
2.10 与其他方法含量测定结果的比较 选取3种维生素预混料、5种复合预混料、1种配合料、1种浓缩料共10批次饲料样品,分别用酶解法(曹冶,1997)、甲醇直接提取法(GB/T 17817-2010 第二法和GB/T 17812-2008第二法)、皂化法(GB/T 17817-2010第一法和GB/T 17812-2008第一法)和本方法处理测定维生素A和维生素E含量,结果如表8。酶解法测定时,不同样品提取效果会有差异,一些样品不能完全被提取,这可能和维生素包被壁材的类型有关系;甲醇提取法测定维生素A普遍偏低,测定维生素E相对较好,只有个别样品偏低,一是因为在甲醇溶剂中,长时间高温使得维生素A产生了异构体,另一方面也可能是和维生素包被壁材的类型有关,一些包膜不能被甲醇破坏溶解,维生素不能完全释出;本方法和皂化法测定结果基本一致,略有偏高(特别是维生素E),这可能是本方法相对较少的步骤和时间,相对较低的皂化温度,优化了皂化体系,减少了维生素的损失。因此在日常饲料检测中可以利用本方法快速测定维生素A和E的含量,结果准确可靠。
3 讨论与结论
目前维生素的壁材包被物质主要有明胶、阿拉伯胶、环糊精、变性淀粉、乳糖、玉米糖浆等以及多种壁材的混合物。淀粉和环糊精在高温和碱的作用下易产生糊化作用分散在含水溶剂中形成胶体溶液;乳糖、玉米糖浆在水中易溶解;明胶、阿拉伯胶和乳清浓缩蛋白等含蛋白质物质在强碱加热条件下容易水解。因此本方法采用皂化法,并且精简优化操作,适用性广,结果更准确,可以广泛应用于饲料中维生素A和维生素E的质量控制。
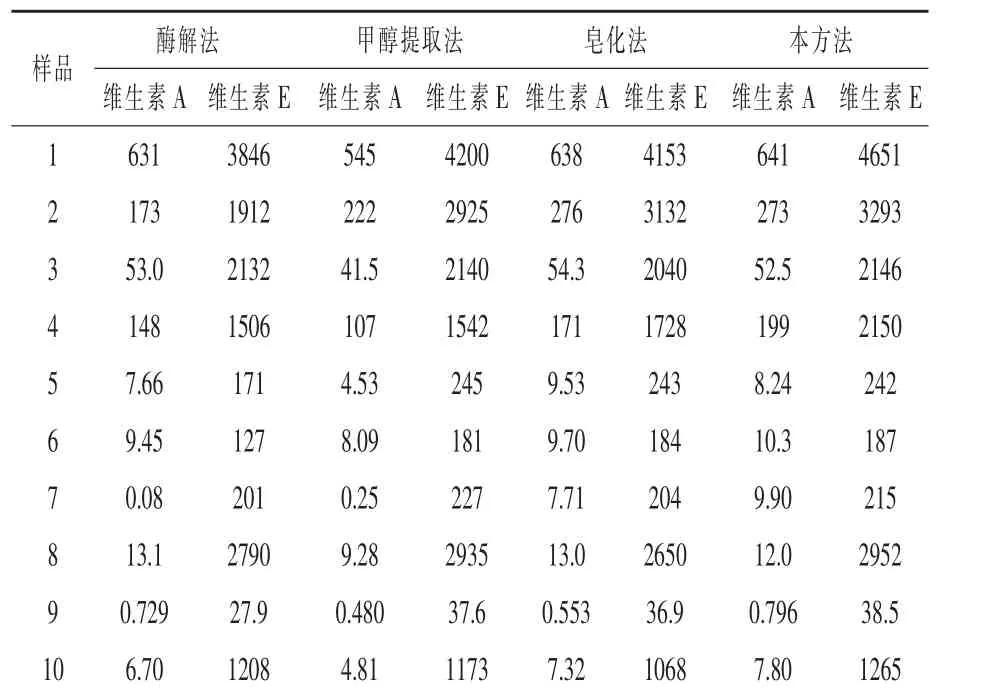
表8 四种方法测定饲料中维生素A和维生素E含量的比较
维生素预混料和复合预混料中主要成分是矿物质或者含纤维素多的物料,不含或含有极少量脂肪和蛋白质,皂化后的产物与其他有机质比较相对单一,采取皂化后直接进行色谱分析,皂化温度相对较低,简化了萃取操作,不仅提高工作效率,还减少样品损失,降低了误差。但是本方法取样量相对国标皂化法少,对于饲料样品的均匀度要求相对较高。
样品提取液溶解有部分醋酸钾等无机盐和其他水溶物,在分析时也会进入到液相色谱系统。特别是其他水溶物,和流动相混合时,容易析出甲醇不溶物,因此在提取时减少盐和水的含量,但又能满足皂化要求,加入碱和抗坏血酸的量都选择较低水平,提取液体系中水的含量控制在10%左右。同时在色谱分析一段时间或者完成全部分析后要用15%左右的甲醇水溶液冲洗色谱柱至少60 min,有利于延长色谱柱的使用寿命。
实际饲料样品中,维生素A含量的范围跨度比较大,使得制备的样品上机液浓度范围跨度也大,在使用相对应宽浓度范围(最高浓度和最低浓度相差2500倍)标准校准曲线进行定量时,高浓度或者低浓度样品的校准浓度和实际浓度有一定的差异,导致定量不准确,因此可以采用高、低两个浓度范围分段做标准校准曲线。
