重组IL-33在致死型约氏疟原虫感染中对细胞因子表达的影响及意义
2019-06-16邢庆洋付俣萧曹雅明
邢庆洋, 付俣萧, 黄 旭, 吕 童, 冯 辉, 曹雅明
(中国医科大学基础医学院 免疫学教研室,辽宁 沈阳 110122)
IL-33属于IL-1细胞因子家族成员,因其在稳态和炎症中的双重作用而受到广泛关注[1]。IL-33既可在细胞外发挥传统细胞因子的调控作用,亦可作为转录因子调控基因表达[2]。IL-33及其受体ST2(suppression of tumorigenicity 2)已被证实参与固有免疫和适应性免疫应答的调节[3]。 疟疾感染诱导产生一系列促炎、抑炎细胞因子和趋化因子,其分泌的时效和程度与疟原虫负荷、感染进程和疾病严重程度密切相关[4-5]。研究发现,重症疟疾患儿血浆IL-33水平明显升高,提示内源性IL-33与疟疾严重性相关[6]。PlasmodiumbergheiANKA(PbA)感染小鼠脑疟发生时,脑组织内IL-33表达上调,相比ST2-/-小鼠脑疟症状得到明显改善,提示内源性IL-33信号参与实验性中枢脑疟(experimental cerebral malaria,ECM)发生[7]。然而,另有研究显示外源重组IL-33对PbA感染小鼠有保护作用。IL-33通过降低炎症因子IFN-γ、IL-12和TNF-α的产生阻止ECM发生[8]。鉴于IL-33在疟疾感染中的双重作用,本研究采用致死型约氏疟原虫(Plasmodiumyoelii17XL,P.y17XL)感染BALB/c小鼠模型,探讨重组IL-33治疗对疟疾感染宿主组织局部细胞因子表达的影响,进而阐述外源性IL-33在疟疾感染中的生物学作用。
1 材料与方法
1.1 材料
1.1.1 实验小鼠 SPF级BALB/c小鼠(8~10周,18~22 g,雌性),购自北京维通利华公司。
1.1.2 试剂与仪器 致死型约氏疟原虫(Plasmodiumyoelii17XL,P.y17XL)由中国医科大学免疫学教研室液氮保种;重组小鼠IL-33(Biolegend,美国);M-PER®蛋白裂解液(Thermo Fisher Scientific,美国);ProcartaPlex®小鼠Th1/Th2细胞因子(9 plex)免疫检测试剂盒(Ebioscience,美国),包括IL-1β、IL-2、IL-4、IL-5、IL-13、IL-6、IL-18、IFN-γ和TNF-α;ProcartaPlex®小鼠趋化因子(9 plex)免疫检测试剂盒(Ebioscience,美国),包括CCL2(MCP-1)、CCL3(MIP-1α)、CCL4(MIP-1β)、CCL5(RANTES)、CCL11(Eotaxin)、CCL21(MIP-2)、CCL7(MCP-3)、CXCL10(IP-10)和GRO-α(CXCL1);酶标仪购自BIO-TEK公司;离心机购自Thermo公司。
1.2 方法
1.2.1 实验模型建立 液氮保种P.y17XL 感染红细胞复苏后,腹腔感染BALB/c小鼠(0.1 mL/只,i.p.)。待原虫血症达10%左右,小鼠麻醉状态下心脏采血EDTA抗凝,获取新鲜P.y17XL 感染红细胞。1×106P.y17XL 感染红细胞腹腔感染BALB/c小鼠(0.1 mL/只,i.p.)。尾静脉血涂片Griemsa染色,计数原虫血症。
1.2.2 IL-33腹腔注射 重组IL-33粉末用PBS溶解,每只小鼠按0.2 μg腹腔注射20 μL,连续给药5 d。重组小鼠IL-33粉末PBS溶解10 μg/mL,0.2 μg/只/200 μL/感染后0 d开始腹腔注射,连续注射5 d。对照组注射同等剂量PBS。
1.2.3 脾细胞培养 无菌获取脾脏,研磨组织获取细胞悬液。离心后获得脾细胞,加入红细胞裂解液4 ℃裂解细胞3~5 min,PBS洗细胞1次。离心获取脾细胞,定容后计数脾细胞总数。M-PER®蛋白裂解液加蛋白酶抑制剂裂解pRBC,获取疟原虫全虫蛋白。96孔细胞培养板10 mg/mL全虫蛋白4 ℃包板过夜,次日无菌接种脾细胞,5% CO2无菌37 ℃培养72 h。收集样本,4 ℃ 1 000×g离心10 min,取上清分装,-80 ℃冻存待用。
1.2.4 多因子免疫检测 血清样本和脾细胞培养上清使用前,4 ℃ 10 000×g离心10 min,取上清。96孔板每孔加入50 μL预混抗体磁珠,放置在手持式磁力板上。150 μL洗脱buffer洗磁珠1次,去掉磁力板。每孔加入样本稀释液(25 μL/孔),再加入血清样本(25 μL/孔)、培养上清样本(50 μL/孔)及标本品(25 μL/孔或50 μL/孔)。密封板后室温500 r/min震荡2 h,洗脱3次,加入检测抗体(25 μL/孔),密封板后室温500 r/min震荡30 min。洗脱3次,加入链霉亲和素-PE(50 μL/孔),密封板后室温500 r/min震荡30 min。洗脱3次,每孔加入120 μL检测buffer,密封板后室温500 r/min震荡5 min。Luminex®200上机检测。ProcartaPlex Analyst 1.0软件分析多因子数据。
2 结果与分析
2.1 IL-33给药组和对照组原虫血症水平
P.y17XL 感染小鼠感染后3 d外周血可见疟原虫感染红细胞。随后,原虫血症水平快速上升,在感染后7 d,感染率达到峰值。与对照组相比,IL-33给药组小鼠原虫血症上升缓慢。感染0~5 d,IL-33给药组原虫血症水平控制在20%以内。IL-33给药组原虫血症峰值比对照组延迟出现(图1)。

图1 IL-33给药组和对照组原虫血症水平
2.2 IL-33给药组和对照组脾细胞培养上清Th1型细胞因子表达水平
感染3 d和5 d,P.y17XL 感染小鼠IL-33给药组和对照组脾细胞培养上清3种Th1型细胞因子检测结果显示,对照组IFN-γ表达水平明显增加(P<0.05,5 d vs 3 d),IL-18在感染后5 d出现明显降低(P<0.01,5 d vs 3 d)。IL-2表达持续处于低水平。与对照组相比,IL-33给药组IFN-γ在感染后3 d或5 d均出现明显降低(P<0.01),IL-18表达明显降低(P<0.05)。在感染5 d,IL-33给药组IL-2和IL-18水平明显低于感染3 d(P<0.05)(图2)。
2.3 IL-33给药组和对照组脾细胞培养上清Th2型细胞因子表达水平
感染3 d和5 d,P.y17XL 感染小鼠IL-33给药组和对照组脾细胞培养上清3种Th2型细胞因子检测结果显示,对照组IL-4、IL-5和IL-13在感染后5 d明显降低(P<0.05,5 d vs 3 d)。与对照组相比,IL-33给药组IL-5在感染后3 d和5 d均明显升高(P<0.05)。IL-13在感染后5 d较对照组明显增加(P<0.05),但比感染后3 d有所降低(P<0.05)(图3)。
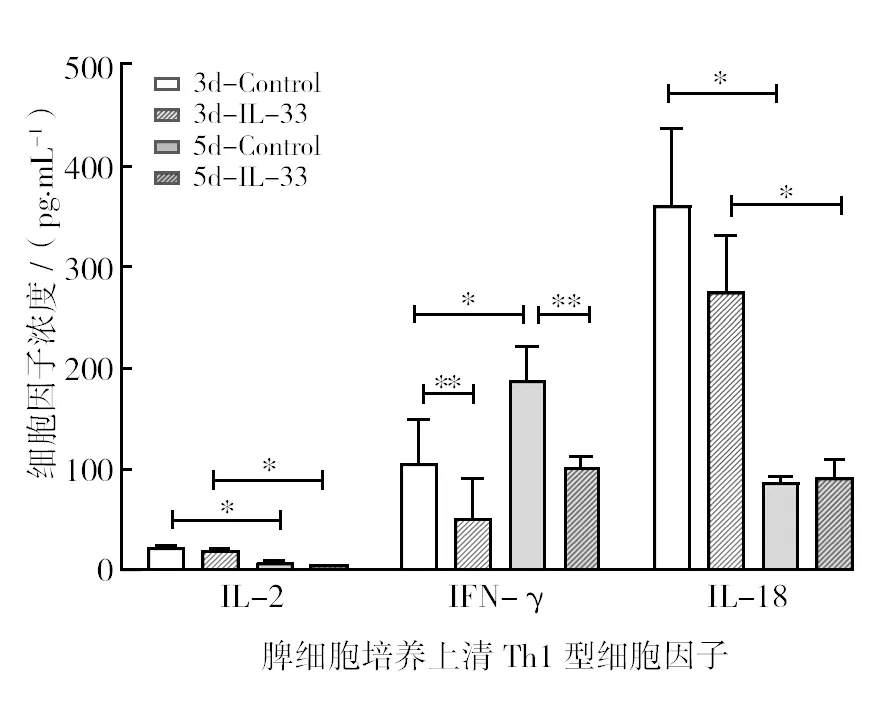
图2 IL-33给药组和对照组脾细胞培养上清Th1型细胞因子表达水平

图3 IL-33给药组和对照组脾细胞培养上清Th2型细胞因子表达水平
2.4 IL-33给药组和对照组脾细胞培养上清前炎性细胞因子表达水平
感染3 d和5 d,P.y17XL 感染小鼠IL-33给药组和对照组脾细胞培养上清3种前炎性细胞因子检测结果显示,与感染后3 d相比,对照组IL-6水平在感染5 d表达明显升高(P<0.05,5 d vs 3 d)。与对照组相比,IL-33给药组IL-6水平在感染3 d和5 d均明显降低(P<0.01)。TNF-α在感染3 d较对照组明显降低(P<0.01)。IL-1β在两组间无明显差别(图4)。

图4 IL-33给药组和对照组脾细胞培养上清前炎性细胞因子表达水平
2.5 IL-33给药组和对照组脾细胞培养上清趋化因子表达水平
感染3 d和5 d,P.y17XL 感染小鼠IL-33给药组和对照组脾细胞培养上清趋化因子检测结果显示,与感染3 d相比,对照组RANTES和IP-10表达水平在感染5 d明显下降(P<0.05,5 d vs 3 d)。与对照组相比,IL-33给药组GRO-α、RANTES在感染3 d均明显降低(P<0.01,P<0.05),IP-10差异无统计学意义。感染5 d,IL-33给药组GRO-α较对照组明显下降(P<0.01)。IL-33给药组组内,感染5 d与3 d相比,RANTES有明显上调(P<0.05),IP-10明显降低(P<0.05)(图5)。
2.6 IL-33给药组和对照组脾细胞培养上清巨噬细胞相关趋化因子表达水平
感染3 d和5 d,P.y17XL 感染小鼠IL-33给药组和对照组脾细胞培养上清趋化因子检测结果显示,与感染3 d相比,对照组MIP-1α表达在感染5 d明显升高(P<0.05,5 d vs 3 d),MIP-1β和MCP-3表达明显降低(P<0.05)。与对照组相比,IL-33给药组MIP-1α、MIP-1β在感染5 d均明显降低(P<0.01,P<0.05),MCP-1和MCP-3无明显差异。IL-33给药组组内,感染5 d与3 d相比,MCP-3有明显下降(P<0.05),Eotaxin和MIP-2几乎无检出(图6)。

图5 IL-33给药组和对照组脾细胞培养上清趋化因子表达水平

图6 IL-33给药组和对照组脾细胞培养上清趋化因子表达水平
3 讨 论
IL-33主要由组织细胞如上皮细胞、成纤维细胞和内皮细胞合成[9]。在稳态状态下,IL-33不被细胞主动分泌。在炎症条件下,IL-33在肥大细胞和树突状细胞等免疫细胞中诱导表达[10]。细胞损伤或死亡是IL-33到达细胞外环境的主要机制。因此,IL-33作为警报素是免疫系统的重要的早期预警[11]。
研究证实,IL-33以全长分子形式(IL-33FL)发挥生物学活性,通过ST2依赖方式诱导靶细胞NFκB活化和细胞因子产生[12-13]。抗血吸虫感染中IL-33可促进嗜酸性粒细胞合成IL-13[14]。ST2+记忆Th2细胞(mTh2)以IL-33依赖方式抑制蠕虫的生殖能力,通过表达IL-5在肺部募集高表达碱性蛋白(MBP)的嗜酸性粒细胞发挥抗蠕虫感染作用[15]。本研究发现,重组IL-33对P.y17XL 感染中多种趋化因子表达下调,其中MIP-1α/β和GRO-α主要趋化单核、巨噬细胞和中性粒细胞,而RANTES对活化T细胞有趋化效应。与此同时,重组IL-33治疗P.y17XL 感染小鼠后脾细胞分泌前炎症细胞因子IFN-γ、IL-6和TNF-α水平明显降低。由此表明,重组IL-33在P.y17XL 感染过程中通过调控趋化因子表达,减少前炎症细胞浸润进而降低前炎症细胞因子表达。
IL-33靶向细胞主要是组成性表达ST2的组织驻留免疫细胞,如肥大细胞、II型固有样淋巴细胞(innnate lymphoid cells,ILC2)和调节性T细胞(Treg)。外源性IL-33促进Pb感染小鼠ILC2扩增、M2巨噬细胞极化和Treg细胞功能增强[8]。在钩虫感染IL-33-/-小鼠中,分泌IL-13的ILC2选择性缺失,嗜酸性粒细胞募集降低,进而不利于虫体的清除[16]。夏氏疟原虫(PlasmodiumchabaudiAS,Pc AS)感染IL-13-/-和ST2-/-小鼠实验结果提示,IL-33/ST2通过产生IL-13诱导BALB/c小鼠产生TNF-α和IL-6等促炎因子,参与PcAS感染的肝脏炎症性损伤[17]。
本研究发现,重组IL-33治疗P.y17XL 感染小鼠后脾细胞分泌IL-5水平明显增加而IL-13变化不明显。嗜酸性粒细胞是主要的IL-5Rα表达细胞,IL-5是组织中嗜酸性粒细胞聚集的主要调节因子[18]。嗜酸性粒细胞通过释放细胞毒性蛋白参与抗寄生虫感染[19]。本研究结果提示,重组IL-33通过上调IL-5表达水平,增强嗜酸性粒细胞活性进而发挥控制P.y17XL原虫增殖的作用。
寄生虫感染中细胞因子和趋化因子表达谱与原虫负荷的相关性研究是寄生虫感染替代标志物的另一种筛查手段[20],促炎和抑炎因子之间的平衡与疟疾低发病率相关[21]。本研究采用多因子检测手段,通过对疟原虫感染状态下脾细胞培养上清中的多种细胞因子和趋化因子进行综合分析,明晰了P.y17XL 感染急性期以炎症性细胞因子和Th1型细胞因子应答为主。重组IL-33治疗可促进IL-5为主的Th2型应答,进而降低Th1和前炎症应答效应,减少炎症损伤。
IL-33在调节免疫细胞和组织应答中具有多重作用,其在日益增多的疾病中的作用越来越受到关注。有关IL-33在体内的生物活性形式,以及如何进入靶细胞产生局部效应或全身性作用尚有待深入研究。随着对IL-33在健康与疾病中的作用方式、调节机制和功能的全面了解,以期对包括疟疾在内的感染性诊断、治疗和预防提供指导与帮助。
