慢性家族性良性天疱疮五家系ATP2C1基因突变分析
2019-06-10崔红宙王德彤郭书萍
王 霆 崔红宙 王德彤 高 杰 郭书萍
慢性家族性良性天疱疮,又名Hailey-Hailey病(Hailey-Hailey disease,HHD,OMIM 169600),是以皮肤褶皱部位反复出现水疱、糜烂为主要特征的常染色体显性遗传性皮肤病[1]。组织病理上以基底层上方棘层松解形成裂隙和水疱为特征。本研究前期收集到典型的HHD 5家系,通过Sanger测序对家系成员进行ATP2C1基因序列检测,现报道如下。
1 对象与方法
1.1 对象 本研究选取来自不同家系的12例患者,最小发病年龄21岁,最大69岁,先证者经我科临床及病理组织检查,确诊为HHD,患者皮损见图1a~1e,家系图见图2a~2e。患者签署知情同意书并收集病史资料及外周血。同时选取13名表型正常的不同家系成员和100名健康对照,签署知情同意书,该研究通过医院伦理委员会审查。
1.2 方法
1.2.1 提取基因组DNA 采集HHD家系中患者及家系内表型正常者的外周静脉血5 mL,存储于2% EDTA抗凝管中,-80℃保存。使用QIA amp DNA blood mini kit(QIAGEN公司,德国)提取基因组DNA,同时以100名与该家系无亲缘关系的健康个体作为对照。
1.2.2 PCR扩增 引物序列设计参考文献[2],由上海生工生物工程技术服务有限公司合成。反应条件:94℃预变性5min,94℃变性30s,64℃退火30s,72℃延伸50s,前12循环退火温度每循环降0.5℃,以58℃为退火温度24个循环,72℃延伸10min。
1.2.3 DNA测序 PCR产物经1.5%琼脂糖凝胶电泳检测后,交与上海生工生物技术服务有限公司行Sanger测序,测序结果使用Chromas软件解读,并与人类基因组数据库ATP2C1基因序列进行对比。

1a:家系1先证者腹股沟、阴囊红斑基础上糜烂、浸渍、裂隙;1b:家系2先证者腋下红斑、浸渍;1c:家系3先证者腋下红斑、浸渍;1d:家系4先证者腹股沟红斑、浸渍;1e:家系5先证者双侧腹股沟、外阴弥漫潮红色斑、糜烂、浸渍
图1先证者临床表现
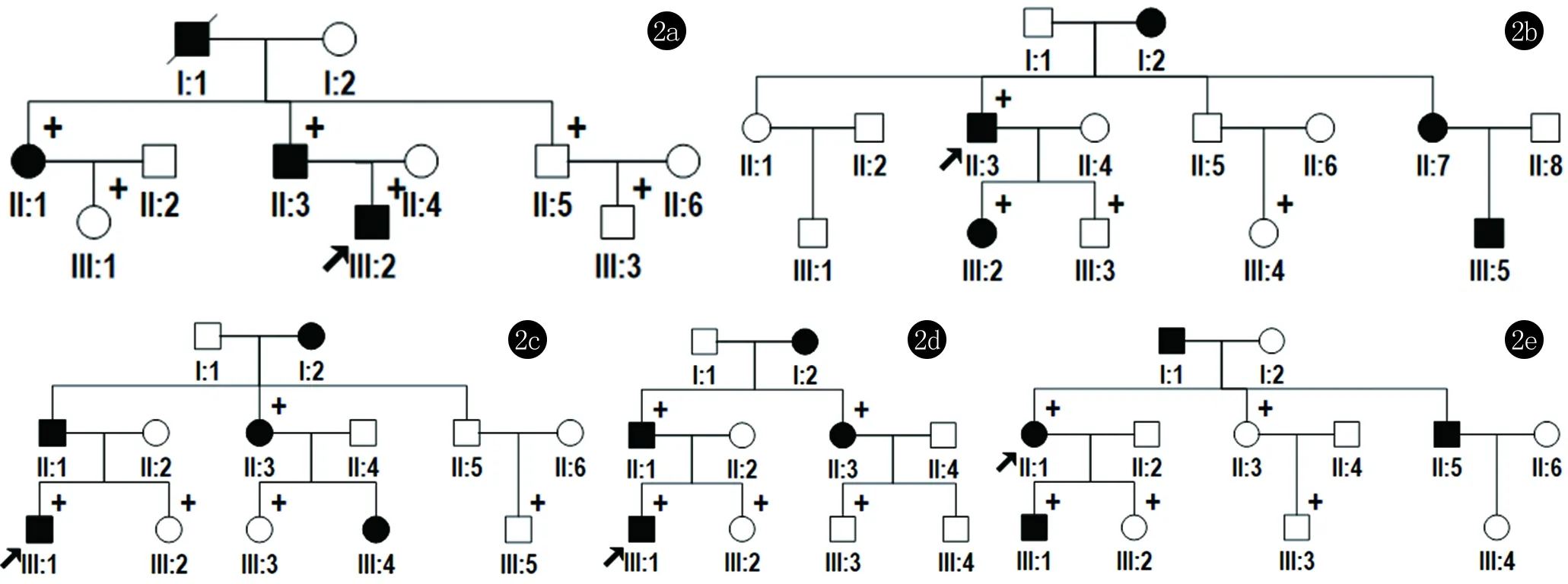
2a~2e分别为家系1~5家系图(标“↑”者为先证者;标“+”者为行ATP2C1基因突变检测)
2 结果
家系1中3例患者在 ATP2C1基因第18号外显子上发现错义突变c.1738A>G(p.I580V)(图3a),使用Polyphen-2软件进行蛋白功能预测,预测结果为possibly damaging (score=0.722)。在表型正常的3名家系成员中未发现该突变;家系2中2例患者在第25号外显子上发现无义突变c.2416C>T(p.R806*)(图3b),2名表型正常的家系成员中未发现该突变。家系3中3例患者在第15号外显子上发现错义突变c.1250G>A(p.R417K)(图3c),使用Polyphen-2软件进行蛋白功能预测,预测结果为benign(score=0.100),同时家系内2名表型正常的个体未发现该突变。这些突变在100名无亲缘关系的正常人中均未发现。家系4、家系5 ATP2C1基因未发现突变。
3 讨论
HHD由Hailey兄弟于1939年首次报道并因此得名。该病呈世界性分布,发病率约为1∶50000,男女发病率大致相同,约70%患者有家族史[1]。该病的临床表现为皮肤褶皱部位反复出现水疱、糜烂,常伴不同程度瘙痒,夏重冬轻。目前尚无有效治疗手段。
1995年,Richard等[2]利用微卫星多态性标记对6个来自德国和意大利的家系行基因连锁分析将HHD致病基因定位于3q21~q24。2000年,Sudbrak和Hu等[3,4]在3q21~q24区域内发现ATP2C1为HHD致病基因。ATP2C1基因包含28个外显子,全长约107.46kb,编码949个氨基酸。截至目前,已发现140余种突变,其中中国汉族人群突变已超过80种[5,6],突变方式包括错义突变、剪切位点突变、无义突变、移码突变及其他方式突变,这些突变散在分布于各个外显子,目前尚无明确的热点突变。人类ATP2C1基因与小鼠SPLA基因和酵母PMR1基因同源[7],均为编码高尔基体介导的钙泵[8]。该基因的最终翻译产物为人高尔基体分泌途径Ca2+/Mn2+ATP酶(hSPCA1)。hSPCA1蛋白在人体多个组织中均表达,其中在角质形成细胞和肾脏内高度表达。这种钙泵可将胞浆内钙离子主动运输至高尔基体内,维持高尔基体内高钙离子浓度和胞浆内低钙浓度,使角质形成细胞对胞外钙离子浓度的变化反应敏感[9]。
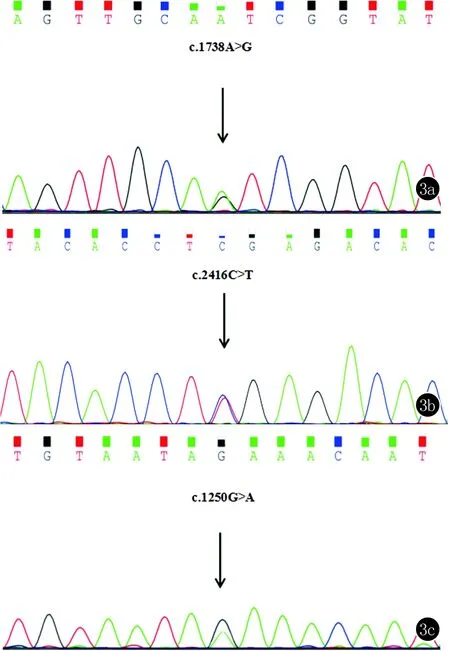
图33a:ATP2C1基因第18号外显子c.1738A>G;3b:ATP2C1基因第25号外显子c.2416C>T;3c:ATP2C1基因第15号外显子c.1250G>A
本研究中发现的c.1738A>G、c.2416C>T、c.1250G>A所编码氨基酸分别位于跨膜片段4、5之间的胞质区和跨膜片段7、8之间的腔道区,我们推测,当胞质区和腔道区发生突变时,必然会影响hSPCA1结构的改变,从而影响其在钙离子或锰离子转运和存储功能,导致细胞内信号转导异常,无法维持高尔基体内钙离子浓度,使其对胞外钙离子变化敏感性降低,从而导致其削弱某些重要蛋白质的糖基化、水解、折叠及转运过程,特别是桥粒糖蛋白、桥斑蛋白的磷酸化和肌动蛋白的合成障碍,最终破坏细胞间黏附[10]。关于家系4、5未发现ATP2C1基因未发现基因突变的原因分析如下:(1)常规PCR扩增、电泳和测序无法检出ATP2C1基因大片段乃至于整个片段的缺失;(2)由于引物设计着重考虑外显子区域而忽略影响剪切、mRNA稳定性及蛋白翻译的内含子、转录的非编码区及启动子区域。
目前大量有关ATP2C1基因突变已陆续被发现,本研究发现的c.1738A>G、c.2416C>T曾于2002年被Dobson-Stone等[10]报道,c.1250G>A曾于2012年在汉族人群中被Zhang等[11]报道。c.2416C>T则是在我国汉族种群首次被报道。对病例进行基因型-表型关联性研究可以促进对基因诊断和精准医疗的发展,下一步我们准备收集更大的样本量,完善中国汉族人群基因型数据库;同时对基因型-表型关联性研究可以更好地指导基因诊断,为HHD患者的精准治疗提供理论基础。
