二芳基乙烯光致变色理论研究
2019-06-05李会学王文笔
李会学,王文笔
(天水师范学院 化学工程与技术学院,甘肃 天水 741001)
二芳基乙烯含有乙烯基团以及连接在两侧的杂芳环基团,二芳基间距离较近,可以形成开环及闭环两种异构体,全氟二芳基乙烯开环状态下为无色,紫外光照后生成闭环异构体,该状态下呈现粉红色,闭环构型在可见光照射下,又可以发生开环反应回到开环构型,实现可逆循环,两个构象室温且光照条件下之间的快速变换,属于光诱导的可逆反应,这是二芳基乙烯光致变色的基本机理。由于不同取代基通常有着不同的物理及化学性能,通过分子修饰可在二芳基乙烯上连接不同取代基,可实现对材料性能的调控。[1-3]全氟二芳基乙烯具有出色的光致变色性能、热稳定性、抗疲劳性以及灵敏度高等优点,已经在变色眼镜、光开关、逻辑门以及信息存储等诸多领域中取得了诸多瞩目的应用。[4-8]但有关其光化学反应机理方面的理论研究还未见报道,本文将采用含时密度泛函对该材料的基态、激发态做构型优化,得到第一激发态势能面上的反应物、产物、过渡态等构型,探讨反应机理。所研究的分子构型如图1所示,闭环化合物为4,5-环己二烯-6,6,7,7,8,8-六氟-2-(4-戊烷苯基)-4,6,7,8-四氢-3aH-茚并[5,4-b]噻吩,开环化合物为4-(3,3,4,4,5,5-六氟-2-苯基环戊烯)-2-(4-戊烷苯基)噻吩。
1 计算方法

图1 二芳基乙烯闭环及开环分子结构
在化学键断裂、分子基态与低激发态能量近简并的情形下,哈特里-福克方法和密度泛函理论不能用单参考态模拟分子性质,而完全活性空间自洽场(CASSF)方法可以在多参考态条件下得到正确的波函数,该方法是研究分子离子以及激发电子态的有效方法,然而活动空间的选择是CASSF计算中的难点,另外由于计算成本的问题,活动空间不可能选择的太大,这也常常导致计算结果与实验不相符的情况。而含时密度泛函(TD-DFT)采用线性响应技术简单地将基态波函数与激发态波函数联系起来,可快速、可靠地获得激发态的势能面,对于许多分子体系,该方法产生的激发能误差较小,激发态的键长误差大约在百分之几,偶极矩和振动频率的误差也在百分之五以内,不仅准确性好,还支持激发态解析梯度运算,同时计算成本也大大降低,[9]已经成为科研工作者研究分子激发态广泛使用的理论方法。通常,光致变色反应只涉及分子的第一激发态,因此本文采用TD-DFT只研究全氟环戊烯在第一激发态的势能面上的反应,所有计算采用Gaussian 09程序进行,杂化密度泛函及基组采用B3LYP/6-31+G(d),计算过程没有对称性限制设置,溶剂化效应采用极化连续模型进行模拟。
2 结果与讨论
2.1 全氟二芳基乙烯在乙腈中的紫外谱图
实验发现开环构型的乙腈溶液其紫外光谱在284nm处有最大吸收峰,[10]而闭环构型的乙腈溶液的紫外光谱在539nm处有最大吸收峰,当用297nm光照射时,开环构型的无色溶液变成粉红色溶液,形成封闭环构型,相反地当用可见光(λ>500nm)照射该粉红色溶液时,又可以完全漂白为无色的开环构型,该全氟二芳基乙烯表现出了良好的光致变色特性,这是由于无色的开环构型和粉红色的闭环构型之间能适时切换。
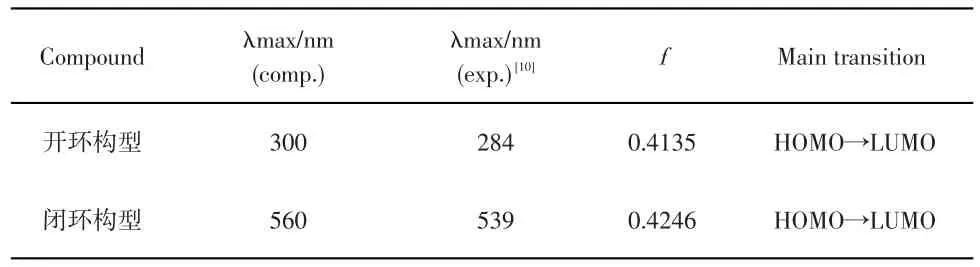
表1 B3LYP/6-31+G*计算得到的二芳基乙烯开环及闭环构型下在乙腈溶液中的最大吸收波长(λmax)、振子强度(f)及跃迁特点(Main transition)
本文采用极化连续模型、B3LYP/6-31+G(d)理论计算了开环及闭环构型的电子光谱,理论预测的最大吸收波长及振子强度列在表1中,所模拟的吸收光谱如图2所示,闭环构型的二芳基乙烯在560nm处出现最大吸收峰,振子强度为0.4246,对应着前线分子轨道上的电子HOMO→LUMO的跃迁,与实验紫外吸收吻合得很好,开环构型的二芳基乙烯在300nm处出现最大吸收峰,振子强度为0.4135,电子跃迁的主要组态仍然对应着HO⁃MO→LUMO的跃迁,与实验紫外光谱图峰值284nm很接近,说明所选的杂化密度泛函是有效的。
2.2 基态势能面的光致变色反应
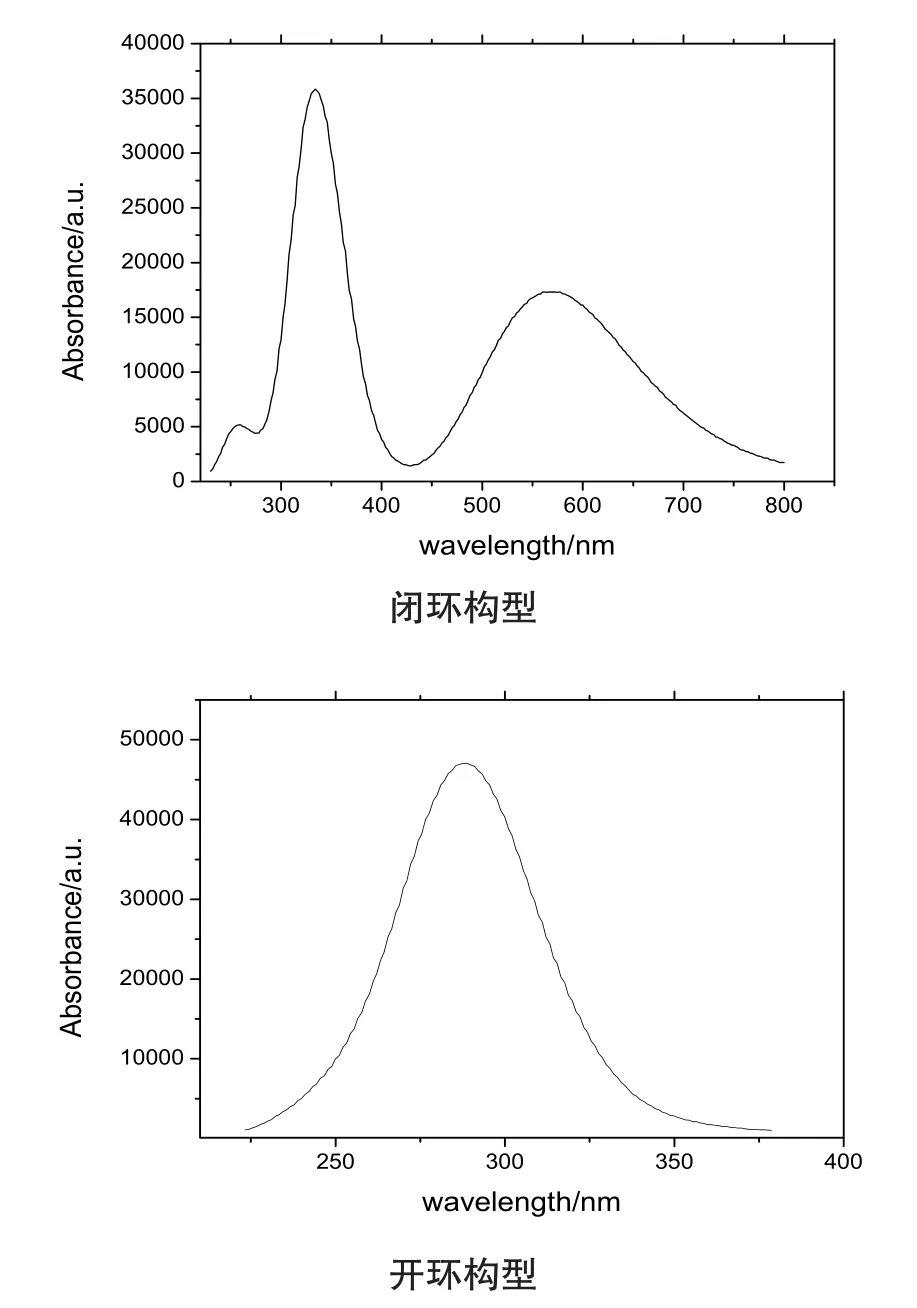
图2 两种构型形成的乙腈溶液紫外光谱理论模拟图
对于二芳基乙烯类有机光致变色体系而言,开环构型能量较低且大都呈现为浅色或无色,而闭环构型为能量较高且呈现出一定颜色,从热力学角度考虑闭环化合物可自发地变为开环构型,但在室温甚至高温下长时间内褪色速率极其缓慢,是所谓的P型热稳定体系,[11]说明要实现快速变色反应在基态条件下很难。本文首先计算了基态势能面上的热化学反应,优化的二芳基乙烯闭环、开环及过渡态构型的部分键参数列于表2,从表2可以看出,开环与闭环部位取决于5C与17C两原子的距离,开环时两原子相距0.3383nm,该距离显然已经超出了化学键成键范围,而闭环构型中两原子相距为0.1526nm,属于典型的C-C单键,该参数表明当两原子靠近时,可有效地形成化学键而生成闭环构型。
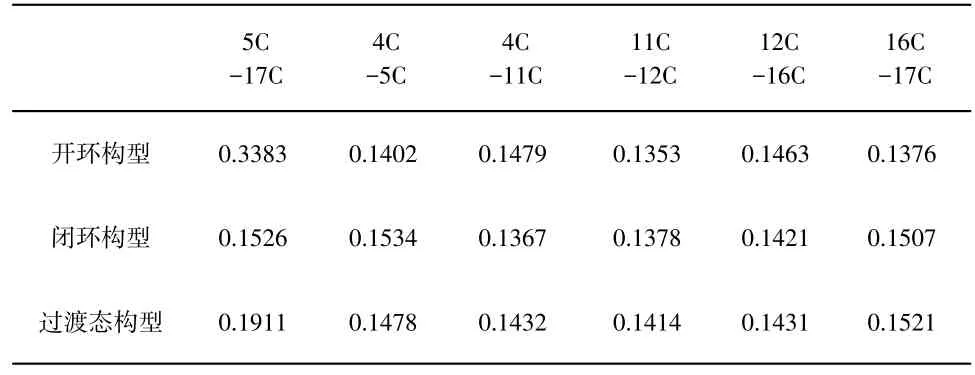
表2 基态下二芳基乙稀开环、闭环及过渡态构型程的部分键参数(nm)
由于知道并优化了反应物和生成物的构型,故采用QST2(Quadratic Synchronous Transit)方法获得该变色反应的过渡态,与反应物、生成物相比,5C-17C的距离介于二者之间,各物种在基态变色反应中势能变化如图3所示,将开环构型的能量作为参考点,即将其内能看作0kJ/mol,则过渡态的相对能量为91.48kJ/mol,生成物能量为45.31 kJ/mol,在基态该变色反应的活化能高达91.48kJ/mol,常温下如没有催化剂的辅助作用,该反应较难发生。
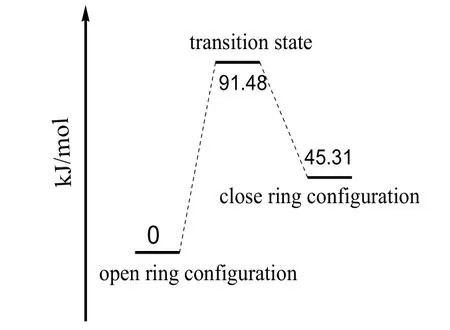
图3 基态全氟二芳基乙烯异构化反应的能量变化
另外,由于内禀反应坐标可以有效、直观地确定最低能量反应路径,我们也计算了该反应的内禀反应坐标(图4所示),获得了沿反应路径的势能剖面,计算结果表明预测的过渡态沿反应通道分别指向反应物与生成物,其唯一虚频是-210cm-1,对应于二面角11C-12C-16C-17C的扭转振动,说明该过渡态是正确的。
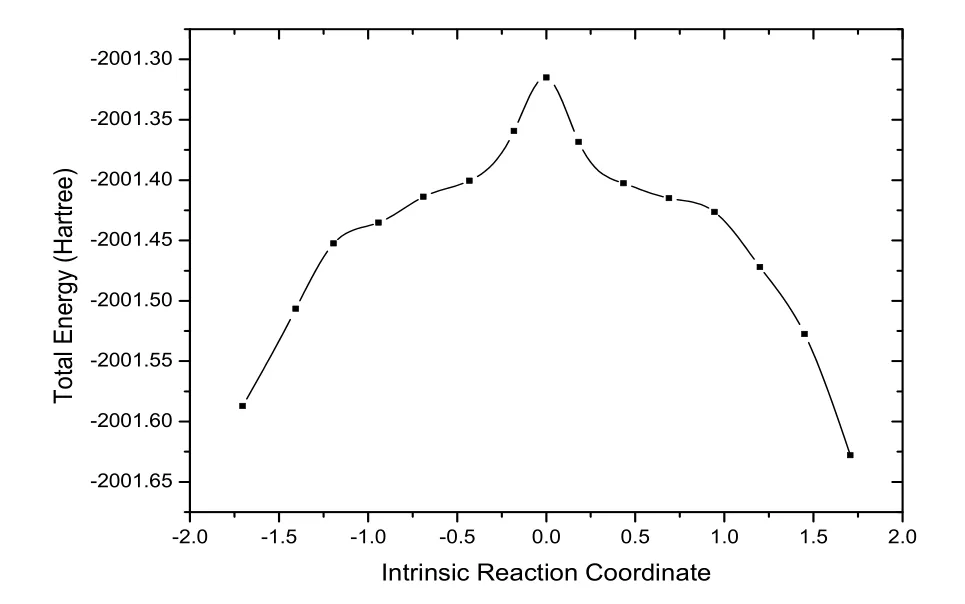
图4 基态势能面的内禀反应坐标
2.2 激发态势能面的光致变色反应
二芳基乙烯的实际应用主要依赖于烯桥和芳基单元的设计选择,本文涉及的化合物其烯桥采用了全氟代的环戊烯,相对于较早设计的强吸电子性的二氰基取代乙烯,具有在光照过程中不会发生顺反异构化反应,抗疲劳程度好,增强了光致变色性能。第一激发态势能面上优化的二芳基乙烯闭环及开环构型的部分键参数列于表3,从表3可以看出,不管是开环还是闭环构型,由于激发态吸收了光子而使得分子内电子重新排布并优化重整,与基态构型的电子密度相比存在差异,故对应的化学键也发生了变化,图5是开环构型与闭环构型的基态及激发态的电子密度差值图,当基态分子吸收光子变为激发态时,电子从蓝色区域迁移到紫色区域,其中激发态闭环构型中5C-17C、4C-11C、11C-12C以及16C-17C等化学键上电子密度降低,键长增大,4C-5C、12C-16C等化学键电子密度增加,故键长减小,激发态开环构型中4C-5C、11C-12C、16C-17C等化学键上电子密度降低,比相应的基态键长要大,化学键4C-11C、16C-17C电子密度增加,这些键长比基态要小。另外计算得到的第一激发态能量变化列于图6中,经计算过渡态能量为21.52 kJ/mol,该数据表明在激发态势能面上反应活化能较小,环境提供一定能量即可使该反应进行,同时计算的产物能量为12.23 kJ/mol,说明当该反应逆向进行时,只需克服9.29 kJ/mol的势垒,故粉红色的闭环构型在可见光照射下即可变为无色的开环构型分子。
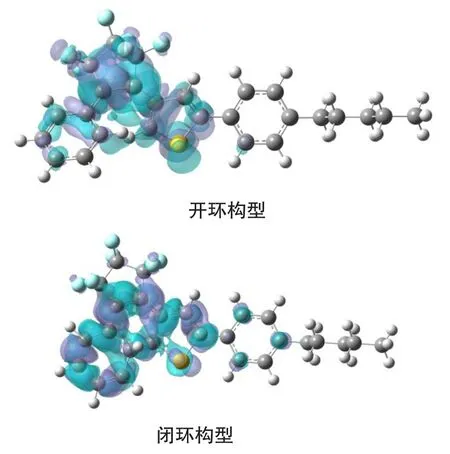
图5 基态及激发态构型的电子密度差值图
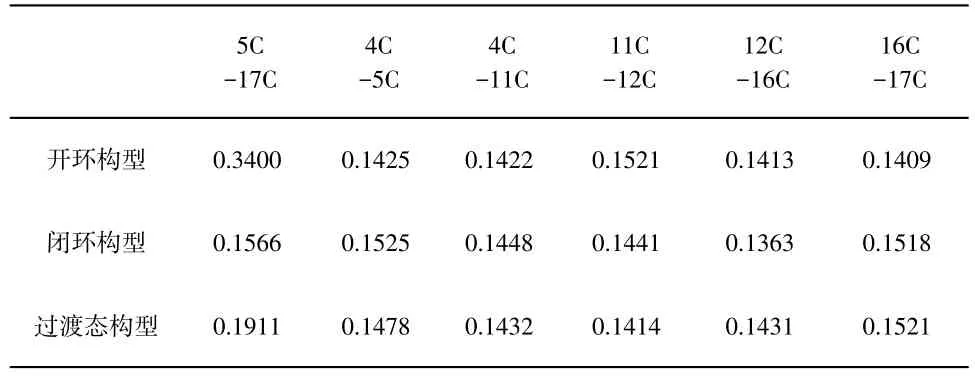
表3 第一激发态下二芳基乙稀开环、闭环及过渡态构型的部分键长(nm)
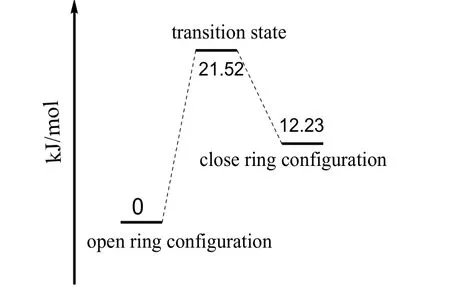
图6 第一激发态全氟二芳基乙烯异构化反应的能量变化
我们也计算了该反应的内禀反应坐标(图7所示),获得了沿反应路径的势能剖面,结果表明预测的过渡态是正确的,其唯一虚频是-150cm-1,对应于二面角11C-12C-16C-17C的扭转振动。

图7 第一激发态势能面的内禀反应坐标
3 结 论
采用杂化密度泛函B3LYP/6-31+G(d)研究了全氟二芳基乙烯光致变色的反应机理,基态条件下由于反应活化能较大,很难发生自发闭环与开环的异构化反应,第一激发态时由于开环及闭环构型得到了活化,反应势垒大为降低,加快了异构化反应的速率,另外还计算了两种构型的紫外吸收光谱,且与实验相符。
