凋亡信号调节激酶-1抑制剂在非酒精性脂肪性肝炎中的研究进展
2019-05-23张小猛雷永华
韩 磊,张小猛,全 旭,雷永华,钱 海
(1中国药科大学新药研究中心,南京 210009;2南京圣和药业股份有限公司,南京 211100)
非酒精性脂肪性肝病(non-alcoholic fatty liver disease,NAFLD)是一种最常见的慢性肝脏疾病,现在全世界约四分之一的成年人患有NAFLD[1],其临床表现为过多的脂肪变性后以甘油三酯的形式堆积在肝脏中[2]。NAFLD的发病机制尚不十分明确,是由环境因素和遗传因素共同导致的,同时,后天的肝脏受损也可能导致肝脏脂肪变性、炎症和纤维化的发生[3]。未经治疗的NAFLD将进展为非酒精性脂肪性肝炎(non-alcoholic steatohepatitis,NASH),并可继续恶化为肝硬化和肝细胞癌(hepatocellular carcinomac,HCC),也是导致终末期肝病以及肝移植的主要原因[4]。
生活方式和习惯的调节,如控制饮食中的热量摄入和增加运动量,目前是NAFLD/NASH治疗的基本手段,但很难改善患者的病情,因此药物治疗显得十分迫切需要,然而,目前仍没有针对该疾病的药物批准上市[5]。
1 现有NASH/NAFLD治疗手段
当前NASH的治疗药物有抗氧化剂/护肝药、过氧化物酶体增殖激活受体(peroxisome proliferator-activated receptor,PPAR)激动剂、降糖药、降脂药、降压药和法尼酯类受体(farnesoid X receptor,FXR)激动剂等[5]。
常用的抗氧化剂/护肝药有维生素E和谷胱甘肽(glutathione,GSH)。维生素E是一种自由基清除剂,可有效降低患者血清谷丙转氨酶(alanine aminotransferase,ALT)和转化生长因子-1活性[6],但长期服用可能诱发前列腺癌[7]和出血性中风[8]。GSH具有抗氧化作用,可降低患者的ALT水平和肝脂肪变性,但其有效性和安全性尚待研究[9]。
吡格列酮(pioglitazone)是常用的PPAR-激动剂,可显著改善患者的脂肪变性和坏死性炎症[10],但可能诱发前列腺癌或胰腺癌以及心血管疾病发病等[5]。胰岛素增敏剂二甲双胍可减少肝糖原生成,增强葡萄糖分解利用,改善肝脏的胰岛素抵抗,降低患者的ALT水平,但并未发现对肝功能和肝组织学的改善[2]。
他汀类降脂药的治疗机制可能与抑制肝脏炎症、改善肝纤维化和降低肝癌的发生有关,但尚未得到证实[11-12]。奥贝胆酸(obeticholic acid,OCA)是一种FXR激动剂,可减少肝脏葡萄糖和脂肪合成同时抑制脂肪变性,降低NASH患者发展为肝纤维化的风险,但会诱导低密度脂蛋白(low density lipoprotein,LDL)水平升高[13]。
上述药物虽有一定的缓解NASH作用,但作用机制不明确且具有明显甚至较严重的不良反应。最新开发的凋亡信号调节激酶-1(apoptosis signal regulating kinases-1,ASK-1)抑制剂(如selonsertib)具有良好的治疗效果,作用机制明确,不良反应较小,现已进入Ⅲ期临床试验阶段,具有良好的发展前景,有望成为首个批准上市的NASH治疗药物,现将ASK-1及其抑制剂的研究进展进行综述。
2 ASK-1的结构及其作用机制
ASK-1是细胞丝裂原活化蛋白激酶激酶激酶(mitogen-activated protein kinase kinase kinases,MAPKKKs)家族成员之一,负责调节c-Jun氨基末端激酶(c-Jun amino terminal kinases,JNK)和p38MAPK通路[3]。当细胞被氧化还原应激以及炎性细胞因子应激时可被激活,从而在天然免疫和病毒感染中发挥重要作用[14-15]。
2.1 ASK-1的结构
ASK-1是一条由大约1 000个氨基酸组成的多肽链,其结构如图1所示。

图1 凋亡信号调节激酶-1(ASK-1)的结构
ASK-1结构的主体部分为上图中的“激酶结构域”部分,而在其的N端和C端两侧各有一个螺旋线圈盘绕结构域,分别称为N端盘管结构域(N-terminal coiled-coil domain,NCC)和C端盘管结构域(C-terminal coiled-coil domain,CCC)[16],这两个区域对于ASK-1单体互相作用形成寡聚体至关重要,而ASK-1在细胞中的活性形式则为寡聚体,也被称为“ASK-1信号小体”[17-18]。
位于ASK-1结构中央的是丝氨酸/苏氨酸激酶结构域,该结构在真核生物中高度保守,包含了ASK-1激活所必须的磷酸化位点,该位点位于激酶结构域的激活环中的Thr845[16],当被激活后引发一系列的蛋白激酶级联反应[19]。
激酶结构域的N端有多个具调节作用的区域,如NCC结构两侧的硫氧还蛋白(thioredoxin,Trx)和TNF受体相关因子(TNF receptor-associated factors,TRAF)特异性结合区域[19]。Trx是一种约12 kD的二硫醇氧化还原蛋白,可以根据细胞的氧化还原状态而改变自身的结构,被认为具有抗凋亡的作用[20]。通常条件下,Trx为还原态形式,结合到Trx结合位点,抑制ASK-1之间形成寡聚体,ASK-1活性被抑制。在氧化还原应激条件下,Trx转化成氧化态形式,与Trx结合位点和激酶结构域之间的区域解离,并且和TRAF蛋白结合,ASK-1的NCC和CCC结构暴露,促进形成寡聚体,进而促进ASK活化和激酶活性[20-21]。TRAF是一种重要的衔接蛋白,对ASK-1下游信号有积极的调节作用[22],在氧化应激情况下与解离下来的Trx结合发挥作用。除此之外,在N端还具有一个CIB1的结合区域,以检测Ca2+应激信号,此外还有其他蛋白-蛋白相互作用的位点[23-24]。
目前对ASK-1的C端区域的研究还不够深入,认为其在ASK-1形成寡聚物的过程中发挥重要作用,且包括一个14-3-3蛋白的结合位点,其对ASK-1活性的影响机制类似于Trx[24-25]。
连接N端Trx结合位点和激酶结构域的是中央调控区,该区域是一个异常致密的四肽重复序列(tetratricopeptide repeat,TPR),并且被一个血小板-白细胞C激酶底物——普列克底物蛋白(pleckstrin)同源结构覆盖,该结构及TPR结构的完整性对ASK-1活性的保持至关重要。相关文献报道[19]提出了其可能的作用机制,其可以变构激活ASK-1的激酶结构域,同时与ASK-1下游底物MKK-6结合,启动MKK-6底物的磷酸化。
通过分析ASK-1与ATP和配体的结合位点,可以了解其独特的结构特征,对于设计选择性ASK-1抑制剂具有重要的意义,图2和图3是文献化合物与ASK-1结合的模式图。
通过文献化合物与ASK-1晶体结构的对接模型分析(如图2所示)表明,ASK-1的关键作用位点位于其铰链区的Val757残基以及内结合口袋位置的Lys709残基[26]。其中,配体咪唑并吡啶结构中咪唑环上的氮原子与Val757骨架的氨基形成氢键,文献化合物紧邻咪唑并吡啶环的氨基与ASK-1主链的羰基形成较强的氢键作用(如图3所示)。配体咪唑环上的氮原子则与ASK-1内结合口袋的Lys709残基的侧链直接相互作用,以上即为ASK-1与配体的3个关键结合位点[26]。除此之外,配体酰胺键的羰基可通过水分子与Ser761形成较弱的氢键作用,叔丁基苯基侧链则伸入ASK-1的溶剂暴露区,此处若连有亲脂性基团则可以占据亲水空间,利于分子间结合[26-27]。由此可见,咪唑并吡啶或其他含有氮原子的母环结构以及连接的酰胺键,含氮杂环侧链均为ASK-1抑制剂设计的关键结构。
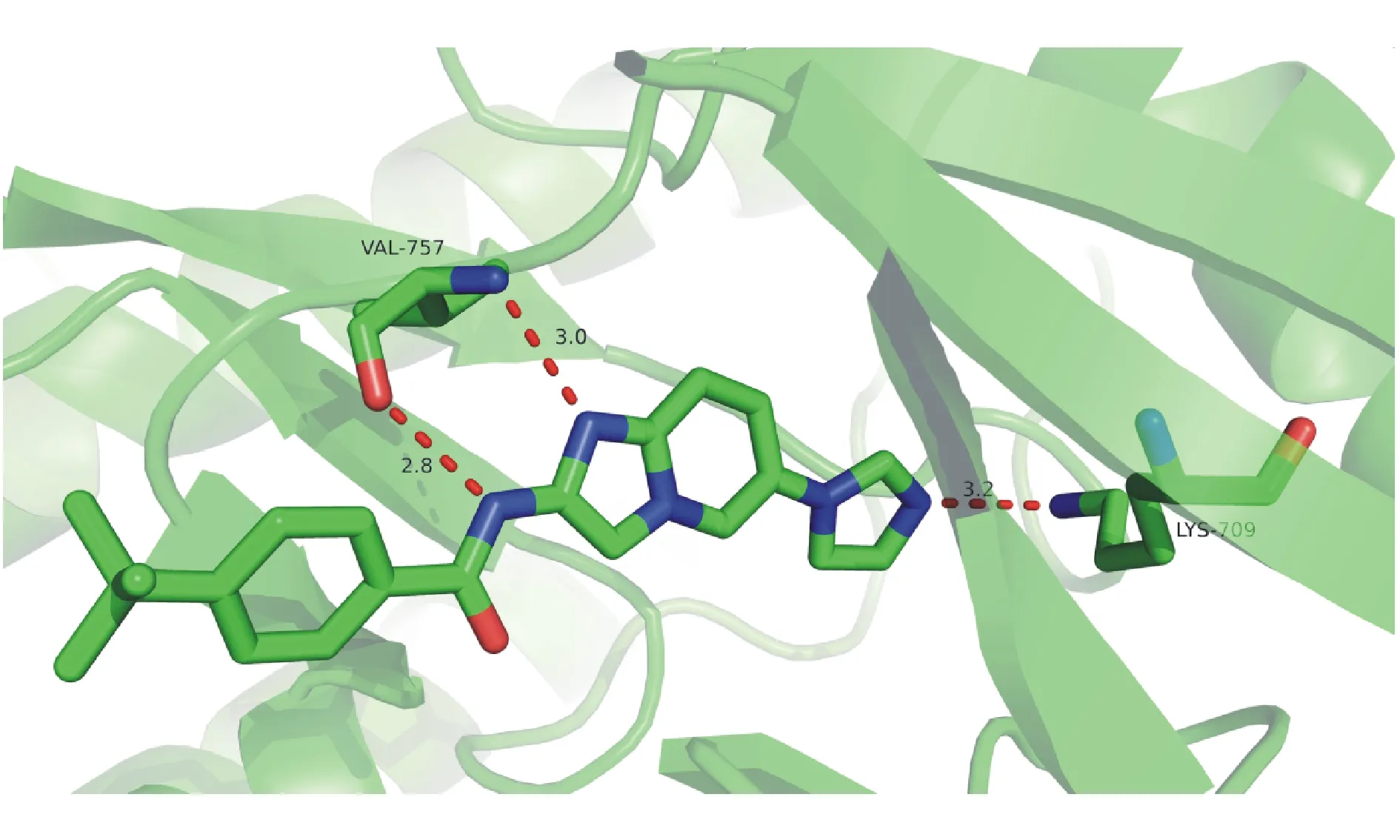
图2ASK-1晶体结构对接模型图(PDB 3VW2)

图3文献化合物与ASK-1结合的化学结构分析图
2.2 ASK-1作用机制
ASK-1的作用机制如图4所示。当机体受到来自多方面的应激刺激时,如细胞内活性氧簇应激(reactive oxygen species,ROS)、内质网应激、钙内流和细胞外炎症信号,如肿瘤坏死因子(tumor necrosis factor,TNF)和脂多糖(lipopolysaccharidec,LPS)等,ASK-1的蛋白激酶级联通路被激活[28]。ASK-1被激活后,Trx从N端解离,ASK-1通过末端寡聚形成ASK-1信号小体,同时,激活结构域激活环内的苏氨酸残基磷酸化,其下游底物MAP2K被激活,此MAP2K底物可分为MKK-4(SEK-1)、MKK-7和MKK-3、MKK-6两条途径。MAPK2K继续磷酸化激活下游的MAPK底物,上述两条途径的底物分别是JNK家族和p38家族[29-30]。激活p38和JNK信号可促进炎症反应、纤维化以及细胞增殖、分化、凋亡,进而诱发多种疾病,如肝脏肾脏疾病、神经退行性疾病、免疫疾病和纤维化等[31]。

图4ASK-1作用机制示意图
因此,ASK-1控制的蛋白激酶级联通路也被称为ASK1-JNK-p38通路,而在NAFLD和NASH患者中,此通路是高度激活的,这种过度激活与胰岛素抵抗、炎症和脂肪增多呈正相关[32]。当ASK-1的活性被抑制时,ASK1-JNK-p38通路被切断,因此导致的细胞增殖、分化、凋亡以及炎症均被抑制。在肝脂肪变性小鼠模型的实验中,ASK-1缺陷型的小鼠通过抑制ASK-1的表达,改善了肝脂肪变性和纤维化程度[33],故ASK-1作为新型的NASH治疗的靶点具有其优越性。
3 ASK-1抑制剂研究现状
当前,ASK-1抑制剂的研究处于蓬勃发展的阶段,全球多家公司都投入了对其的研发,其中首个进入临床研究的是吉利德(Gilead)公司的selonsertib(GS-4997,1),也是当前进展最快且最有希望获得上市许可的化合物。由于selonsertib表现出较强ASK-1抑制活性和对ASK-1相关疾病的显著治疗效果,本文将介绍selonsertib的研究进展以及其他在研治疗NASH已报道的化合物。
3.1 selonsertib
吉利德公司于2013年8月首先公布了ASK-1抑制剂selonsertib的专利文献。Selonsertib首先被用于治疗肺动脉高压和糖尿病肾病,但因其Ⅱ期临床结果不理想(NCT02234141和NCT02177786)进而转向对NASH治疗的研究,并取得了较为满意的治疗效果,目前已进入Ⅲ期临床阶段。

该化合物具有与ASK-1的ATP结合位点竞争性结合的酰胺键和三氮唑结构,通过多项生物学分析也证实其为一个强效的ASK-1抑制剂。经时间分辨荧光能量共振转移(TR-FRET)测定法测得selonsertib抑制ASK-1的IC50为3.0 nmol/L;ASK-1的293细胞水平测定得到其在细胞中的效能EC50为2.0 nmol/L;Kd为0.24 nmol/L,能够在缺少ATP的情况下有效结合ASK-1受体;在人血浆中的未结合分析物百分比为11.94%;经药物代谢动力学测试得到t1/2为5.07 h,口服生物利用度为75%[34]。这些测试中selonsertib均表现出优秀的ASK-1抑制活性和良好的耐受性,在同期吉利德所设计的化合物当中脱颖而出,成为吉利德公司主推的化合物。同时,在小鼠的NASH模型中,selonsertib也显著改善了NASH相关代谢参数以及肝脂肪变性、炎症和肝纤维化程度[35]。
2015年6月,selonsertib进入Ⅱ期临床试验,以研究其单用或联合simtuzumab(SIM)治疗NASH和2期至3期肝纤维化的安全性、耐受性和有效性(NCT02466516)。SIM是一种人源化单克隆抗体,可抑制类赖氨酸氧化酶分子-2(lysyl oxidase-like molecule 2,LOXL2)[36],与ASK-1抑制剂联用可抑制纤维化[37]。此项试验将72名受试者分为selonsertib 6 mg或联用SIM、selonsertib 18 mg或联合使用SIM以及单用SIM 3个大组,部分试验结果如表1所示[38]。
表1Selonsertib Ⅱ期临床试验部分结果

试验指标 Selonsertib 18 mg±SIM(例数/总数)Selonsertib 6 mg±SIM(例数/总数)SIM(例数/总数)纤维化改善≥143%(13/30)30%(8/27)20%(2/10)转变为肝硬化3%(1/30)7%(2/27)20%(2/10)肝僵硬程度削减≥15%15%(4/26)32%(7/22)0%(0/7)肝脂肪减少≥30%26%(8/31)13%(3/24)10%(1/10)
SIM:Simtuzumab,一种LOXL2抑制剂,与ASK-1抑制剂联用可抑制纤维化
在安全性评价方面,部分患者治疗过程中出现至少一次不良反应,但多为轻度症状,主要表现为头痛、恶心、鼻窦炎、鼻咽炎、上腹痛、背痛和疲劳,仅有3例因较严重的不良反应而中断治疗[38]。
Ⅱ期临床试验的结果表明,selonsertib对于NASH和中度至重度的肝纤维化患者具有明显的疗效,且该疗效具有一定的剂量依赖性,但也存在一定不良反应的风险,还需要通过更进一步的研究以阐明其毒理机制,扩大治疗窗。对此,吉利德公司于2016年末和2017年初开启了selonsertib的Ⅲ期临床试验,以研究selonsertib治疗NASH和桥接纤维化以及NASH代偿性肝硬化的安全性和有效性(NCT03053050和NCT03053063),目前仍在进行中。2015年1月15日selonsertib获得了“孤儿药候选药物认证”,是当前在研ASK-1抑制剂中最有希望获得上市和临床实际应用的产品,具有良好的开发前景。
3.2 其他在研的ASK-1抑制剂
除selonsertib外,目前尚没有其他进入临床试验阶段的ASK-1抑制剂,但近期全球多家制药公司均以selonsertib作为参照化合物设计合成了新化合物,保留其母环结构进行了结构修饰和改造,且生物学检测显示具有较强的ASK-1抑制活性和较好的药物代谢动力学性质,部分化合物较selonsertib更加具有优势,具有良好的应用前景,部分化合物的结构和生物学检测结果如表2所示[39-42]。
表2其他在研(生物学测试)的ASK-1抑制剂

序 号结 构IC50/(nmol/L)研发公司20.46东阳光30.28东阳光40.43东阳光50.46东阳光60.9±0.1科伦豪泰70.91±0.12科伦豪泰

(续表)
表2所示的是部分已经通过专利公布的ASK-1抑制剂的化学结构,且具有较好的ASK-1抑制活性,根据对ASK-1与配体结合的晶体结构图分析,与ASK-1铰链区Val757残基和内结合口袋Lys709残基相互作用的酰胺以及临近含氮杂环和侧链含氮杂环(通常是咪唑或三氮唑)是抑制剂设计的必需基团。
其中,化合物2~5为广东东阳光药业于2018年5月公开的专利所报道的化合物,并使用酶法测定对ASK-1的抑制活性,筛选出了上述4个IC50低于1 nmol/L的化合物;此外,经过雄性SD大鼠的药代动力学测试,所筛选的化合物经口给药后在大鼠体内血药浓度及暴露量水平较高,清除率较低,具有良好的生物利用度;药物组织分布的研究发现化合物在大鼠肝和肾器官中的浓度较高[39],可针对性抑制肝脏ASK-1活性,有发展成为NASH药物的潜力。
化合物6~8为四川科伦豪泰生物医药于2018年8月23日公开的专利所报道的化合物,经酶法筛选出上述IC50低于1 nmol/L,对ASK-1酶具有较强的抑制活性;构建pcDNA3.1-ASK1表达载体测定化合物对细胞ASK-1酶的作用,表现出良好的抑制作用;药物代谢动力学实验也表现出良好的生物利用度[40]。
礼来公司2018年9月7日公开的专利披露了其在研的ASK-1抑制剂的化学结构,其将selonsertib酰胺键右侧的结构完全保持,苯环或吡啶环与临近酰胺键成环,其余的改动则集中在苯环或吡啶环的取代基上。通过ASK-1酶实验和ASK-1自磷酸化实验测试ASK-1活性筛选出具有较好抑制活性的化合物9和化合物10[41]。
化合物11~13为江苏豪森药业于2018年9月7日公开的专利所报道的部分化合物。豪森药业通过ASK-1酶学实验TR-FRET法筛选出激酶酶活抑制的IC50小于1 nmol/L的数个化合物,其作为ASK-1的有效抑制剂对NASH的治疗具有巨大的应用潜力。随后进行药代动力学分析,筛选出上述同时具有良好代谢性质、暴露量和最大血药浓度的化合物,在体内的有效持续时间可达3 h以上,其半衰期较selonsertib也有明显的延长。此外还进行了化合物对高脂饮食和CCl4诱导的NASH模型小鼠血清中ALT和AST水平的影响实验,与selonsertib 对比发现,所筛选的化合物均在下调NASH小鼠血清中ALT和AST的水平中表现出良好的效果,明显强于selonsertib[42]。
4 总结和展望
非酒精性脂肪性肝炎作为发病率最高最常见的慢性肝脏疾病,目前临床治疗中除通过日常生活饮食习惯的调节外,推荐的药物治疗方案疗效不明显、不良反应大、给药途径不够友好,使得药物科学家和患者迫切需要更加理想的治疗药物。ASK-1是近年来受到广泛关注的新型靶点,其靶点作用机制清晰,同时,与配体的结合晶体结构模型分析的深入对抑制剂的设计提供了有价值的参考信息,多家制药公司以及科研机构投入对其的研究。其中selonsertib成为进展最快,最有希望获得上市的药物,在Ⅰ期临床和Ⅱ期临床中表现出良好的ASK-1抑制活性和对NASH病人各项指标的有效缓解,具有较好的安全性,当前已进入Ⅲ期临床试验阶段,将进一步对药物有效性和安全性进行评价。各公司以selonsertib为先导所设计的化合物也在临床前的生物学测试中表现处良好的ASK-1抑制活性和药代动力学性质,其中豪森公司还实施了化合物对小鼠血清中ALT和AST水平影响的实验,可以明显降低其水平,具有继续推进的潜力。随着对ASK-1抑制剂的研究更加深入,会有更加有效且不良反应更小的化合物被发现,以ASK-1为靶点的药物有望为NASH患者带来福音。
