超高效液相色谱-串联质谱法同时快速测定食用油中9种外源性杂质成分
2019-05-22罗辉泰黄晓兰吴惠勤张秋炎朱志鑫林晓珊马叶芬
罗辉泰,黄晓兰,吴惠勤,张秋炎,朱志鑫,黄 芳,林晓珊,马叶芬,邓 欣
(广东省测试分析研究所 广东省化学危害应急检测技术重点实验室 广东省中药质量安全工程技术研究中心,广东 广州 510070)
我国是世界上植物油产量和消费量最大的国家[1],废弃油脂量呈逐年上升,部分不法商贩在利益驱动下,将各种来源的废弃劣质食用油(俗称“地沟油”)经脱色、精炼后作为“正常”食用油销售,严重威胁人们的身体健康。我国政府高度重视地沟油非法回流到百姓餐桌的问题,国家卫生部、质监部门大大加强了监管力度。但由于地沟油的鉴别难度极大,至今仍未发布标准检测方法,对地沟油也无明确的认定标准,导致政府部门难以进行有效监管。因此,研究建立指标有效、方法可靠的非正常来源食用油的鉴别方法,对于打击不法分子的上述恶劣行径、保障广大人民的食品安全具有重要意义。
近年来,国内外学者研究建立了测定油脂的电导率、脂肪酸组成、胆固醇等常规理化指标的鉴别方法[2-7],但这些方法均存在专一性不强或灵敏度低等不足,很难准确鉴别地沟油。吴惠勤等[8]提出通过检测食用油中痕量外源性杂质成分来鉴别地沟油,研究建立了测定易挥发外源性杂质的气相色谱-质谱法[8-10]以及部分难挥发外源性杂质的液相色谱-串联质谱法[11],用于地沟油的鉴别,取得了较满意的结果。其他学者也研究了地沟油中的少数几种(2~4种)难挥发外源性杂质[12-15],但种类覆盖面小,易漏检。本文在原有研究基础上[11],针对食用油在烹饪过程中与调味品接触而引入的杂质成分,采用超高效液相色谱-串联质谱(UHPLC-MS/MS)建立了同时测定食用油中来源于4类调料的9种难挥发外源性杂质成分的方法,这些特征成分包括辣椒素、二氢辣椒素、合成辣椒素、胡椒碱、四氢胡椒碱、6-姜酚、6-姜烯酚、10-姜酚和羟基-α-山椒素。本方法样品处理简便快速、成本低、灵敏度高、定性可靠、定量准确,为非正常来源食用油的鉴别提供了可靠方法,可为政府相关部门建立标准方法提供科学依据和技术借鉴。
1 实验部分
1.1 仪器与试剂
Agilent 1290ⅡUHPLC/6470A Triple Quard MS超高效液相色谱-串联四极杆质谱联用仪(美国Agilent公司);赛多利斯TP-114电子天平(美国Sartorious公司);XW-80A快速混匀器(海门市麒麟医用仪器厂);H1850离心机(湖南湘仪实验室仪器开发有限公司);DC12H水浴式氮吹浓缩仪(上海安谱科学仪器有限公司)。甲醇、乙腈均为色谱纯(德国Merck公司);甲酸、乙酸、甲酸铵及乙酸铵均为LC-MS级(美国Sigma公司);实验用水为二次蒸馏水,其余试剂均为分析纯。
9种对照品及内标物:辣椒素、二氢辣椒素、胡椒碱、6-姜酚(纯度均为98%)购自中国食品药品检定研究院;羟基-α-山椒素、10-姜酚、6-姜烯酚和合成辣椒素(纯度均为98%)购自成都普瑞法科技开发有限公司;四氢胡椒碱(纯度97.1%)购自成都瑞芬思生物科技有限公司;内标物苯基辣椒碱(纯度98%),来自某委托送检客户提供的高纯度对照品。
1.2 标准溶液的配制
分别准确称取对照品及内标物各20.0 mg(精确至0.01 mg)至10 mL容量瓶,用甲醇溶解并定容至刻度,混匀得到2 000 mg/L的单标储备液,置于棕色瓶中,于-20 ℃保存。取内标单标储备液适量,用丙酮配制成质量浓度为500 ng/mL的内标工作溶液,置于棕色瓶中,于4 ℃保存;根据需要,取各待测物的单标储备液适量,用乙腈稀释成所需浓度的混合标准工作液。
1.3 样品预处理
准确称取均匀试样1.0 g(精确至0.01 g),置于10 mL聚四氟乙烯离心管中,加入50 μL内标工作溶液(质量浓度为500 ng/mL),充分涡旋混匀,加入8 mL乙腈,涡旋提取3 min,10 000 r/min 离心5 min,将上清液全部转移至10 mL试管;向残液中加入5 mL乙腈,重复提取1次,10 000 r/min 离心5 min,合并上清液,于50 ℃水浴中氮吹浓缩至近干,准确加入1 mL乙腈复溶,超声30 s,充分涡旋混匀,于-20 ℃冷冻3 h,取上清液过0.22 μm滤膜后,待测定。
1.4 色谱-串联质谱条件
1.4.1 色谱条件色谱柱:XSelect®HSS PFP(100 mm×2.1 mm,3.5 μm,美国Waters公司);流动相:A相为5 mmol/L甲酸铵,B相为乙腈;梯度洗脱程序:0~5.50 min,35%B;5.50~6.00 min,35%~40%B;6.00~7.50 min,40%~65%B;7.50~7.51 min,65%~100%B;7.51~8.50 min,100%B;8.50~8.51 min,100%~35%B;8.51~10.00 min,35%B。流速:0.45 mL/min;进样量:5 μL;柱温:30 ℃;分析时间:10 min。通过六通切换阀控制色谱柱流出液流向:在2.6~8.6 min之间,将流出液切换至质谱仪进行分析,其余时间的流出液均被切换至废液中。
1.4.2 质谱条件离子源:安捷伦喷射流电喷雾离子(AJS ESI)源;扫描模式:正离子;采集方式:多反应监测(MRM);干燥气(N2)温度:325 ℃;干燥气(N2)流量:10.0 L/min;雾化气(N2)压力:310 kPa(45 psi);鞘流气(N2)温度:325 ℃;鞘流气(N2)流量:11.0 L/min;毛细管入口端电压:4 000 V;喷嘴电压:500 V;驻留时间:25 ms;碰撞池加速电压:5 V;优化后的质谱采集参数见表1。

表1 9种待测物和内标物的分子式、保留时间及质谱采集参数Table 1 Molecular formula,retention times and MS parameters of nine analytes and the internal standard
tR:retention time;CE:collision energy;*quantitative ion;**internal standard
2 结果与讨论
2.1 非正常来源食用油中外源性特征杂质成分的确定
食用油与非正常来源食用油的主要成分均为脂肪酸甘油酯。油脂本身含有的微量成分,可称为内源性微量成分;而外源性杂质成分,是指油脂在烹饪过程中与食物、调料等接触,以及在回收、储存和加工过程中与包装物等接触而引入的微量成分。作者在前期研究[11]中建立了食用油中辣椒素、二氢辣椒素和胡椒碱等外源性杂质成分的检测方法。本文经进一步研究,总结人们日常生活中的常用调料,将辣椒及其制品中的主要特征成分辣椒素、二氢辣椒素和合成辣椒素,胡椒类调料中的主要特征成分胡椒碱和四氢胡椒碱,姜类调料中的主要特征成分6-姜酚、6-姜烯酚和10-姜酚,以及花椒类调料中的主要特征成分羟基-α-山椒素等9种化合物作为食用油的外源性特征杂质成分。上述成分不存在于正常来源的食用油中,因此可作为鉴别部分食用油来源正常与否的可靠指标。
2.2 质谱条件的优化
从化学结构上看,胡椒碱、羟基-α-山椒素和四氢胡椒碱为碱性化合物,辣椒素、二氢辣椒素和合成辣椒素均为带酚羟基和含氮官能团的两性化合物,6-姜酚、6-姜烯酚和10-姜酚均为含酚羟基的酸性化合物,而9种待测物均适于采用电喷雾离子源测定。
在电喷雾离子源的正、负离子模式下,分别对9种待测物标准溶液作一级质谱全扫描分析,比较了挥发性有机酸(甲酸和乙酸)及其铵盐(甲酸铵和乙酸铵)对化合物离子化的影响。结果表明,胡椒碱等碱性化合物适合在正离子模式下测定,获得准分子离子[M+H]+和加合离子[M+Na]+,且加合离子在挥发性有机酸存在下响应极高,而使用铵盐时丰度显著降低;辣椒素等两性化合物在正、负离子模式下,分别获得准分子离子[M+H]+和[M-H]-,以及[M+Na]+、[M+K]+和[M+Cl]-等加合离子,但正离子的响应明显优于负离子,且使用铵盐亦会抑制正离子模式下加合离子的生成;6-姜酚和10-姜酚等姜辣素在使用铵盐时,获得[M+H-H2O]+和[M-H]-两个基峰离子以及丰度比低于10%的[M+NH4]+、[M+Na]+、[M+H]+和[M+Cl]-等离子,而使用挥发性有机酸时,正离子基峰为[M+Na]+,负离子响应下降明显;姜烯酚类化合物6-姜烯酚在上述4种流动相添加剂条件下,正离子基峰均为[M+H]+。姜辣素和姜烯酚类化合物的实验结果与文献[16]报道一致。实验最终选择[M+H-H2O]+作为6-姜酚和10-姜酚的母离子,[M+H]+作为其余7种待测物的母离子。
另外,某些化合物极易发生源内碰撞诱导解离(Collision-induced dissociation,CID),如合成辣椒素易碎裂生成m/z122.1、m/z137.1等离子,6-姜烯酚、6-姜酚和10-姜酚等易碎裂生成m/z122.1、m/z137.1、m/z177.1、m/z117.1、m/z145.1及m/z149.1等离子,通过进一步优化毛细管出口端电压(Fragmentor),可减少源内CID,使得各化合物的母离子响应最佳。然后对母离子作子离子全扫描分析,根据欧盟2006/657/EC决议中有关质谱分析不得少于4个识别点的规定[17],选取丰度较大的2个子离子作为特征碎片离子,通过MRM模式优化碰撞能量使其响应最佳。优化后的质谱采集参数见表1。最终根据色谱保留时间和2对MRM离子对的丰度比对各组分进行定性鉴别,以MRM定量离子对的峰面积按内标法进行定量测定。
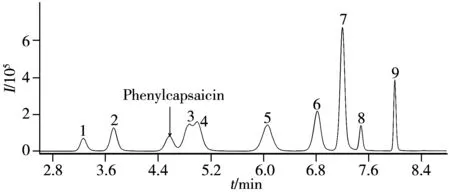
图1 20 μg/L混合标准溶液的总离子流(TIC)色谱图Fig.1 Total ion current(TIC) chromatogram of mixed standard solution(20 μg/L)peak numbers are the same as those in Table 1
2.3 色谱条件的优化
2.3.1 色谱柱的选择由9种待测物的化学结构可知,其极性均为中等偏小,适合采用反相色谱分析。另外,为实现其中结构相近的几组同类成分的良好分离,对比了具有不同选择性的色谱柱Poroshell 120 HPH-C18(100 mm×2.1 mm,2.7 μm)(A)、Poroshell 120 Bonus-RP(100 mm×2.1 mm,2.7 μm)(B)、XSelect®HSS PFP(100 mm×2.1 mm,3.5 μm)(C)和Poroshell 120 PFP(100 mm×2.1 mm,2.7 μm)(D)的分离效果。结果表明:色谱柱A、B和D均未能实现合成辣椒素和辣椒素、胡椒碱和四氢胡椒碱的良好分离,且不能有效分离胡椒碱(m/z286.2)的干扰离子对;而色谱柱C对9种待测物及胡椒碱的干扰离子均有良好的分离,除合成辣椒素和辣椒素外,其余成分均实现基线分离(见图1),因此选择XSelect®HSS PFP(100 mm×2.1 mm,3.5 μm)作为色谱分析柱。
2.3.2 流动相的选择结合质谱条件的优化结果,采用选定的色谱柱,分别考察了以乙腈或甲醇为有机相,5 mmol/L甲酸铵或5 mmol/L乙酸铵为水相时的色谱分离情况。结果表明,上述两种水相对待测物的分离效果无明显差异,但使用甲酸铵时化合物的质谱响应略高;以甲醇为有机相需要延长分析时间来改善色谱分离度。综合考虑,选择5 mmol/L甲酸铵为水相,乙腈为有机相,经反复试验确定“1.4.1”所述的梯度洗脱程序,可在10 min内获得良好的分离效果。图2为9种待测物混合标准溶液的MRM色谱图。
2.4 样品前处理条件的优化
食用油中主要成分脂肪酸甘油酯的极性较小,易溶于正己烷等弱极性有机溶剂,而9种待测物的极性也为中等偏弱,因此提取难度较大。为有效提取目标物和减少油脂带出,选取极性较大的甲醇、乙醇、乙腈等有机溶剂进行试验。结果表明,3种溶剂均能高效提取食用油中的9种待测物,提取率相当,但对油脂的带出量,则乙醇最多,甲醇次之,乙腈最少。因此选用乙腈为提取溶剂。此外,尝试使用乙腈饱和正己烷进行除脂,有一定净化效果,但目标物损失严重。最终选用低温冷冻的方式,可减少样液中的部分油脂,获得较理想的提取效果。
2.5 基质效应的考察
与其他分析方法不同,质谱分析尤其是液相色谱-质谱分析方法通常存在基质效应(Matrix effect,ME)[18],影响方法精密度和准确度。本研究采用提取后添加的方式考察各待测物的绝对基质效应,即采用纯溶剂乙腈和空白基质溶液为稀释剂,分别配制6个质量浓度的混合标准溶液上机测定,按公式ME=B/A计算(B为基质匹配标准曲线的斜率,A为纯溶剂配制标准曲线的斜率),ME比值越接近1,说明基质效应越小,反之亦然。由表2可知,6-姜酚、6-姜烯酚和10-姜酚存在较大的基质效应(ME<0.8),其余待测物均无明显的基质效应(ME值均接近1)。因此,为消除或补偿基质效应给定量带来的偏差,本法选择食用油中不存在、人工合成的作为船舶防污用漆的辣椒碱类似物——苯基辣椒碱为内标物,采用内标法进行定量校正。
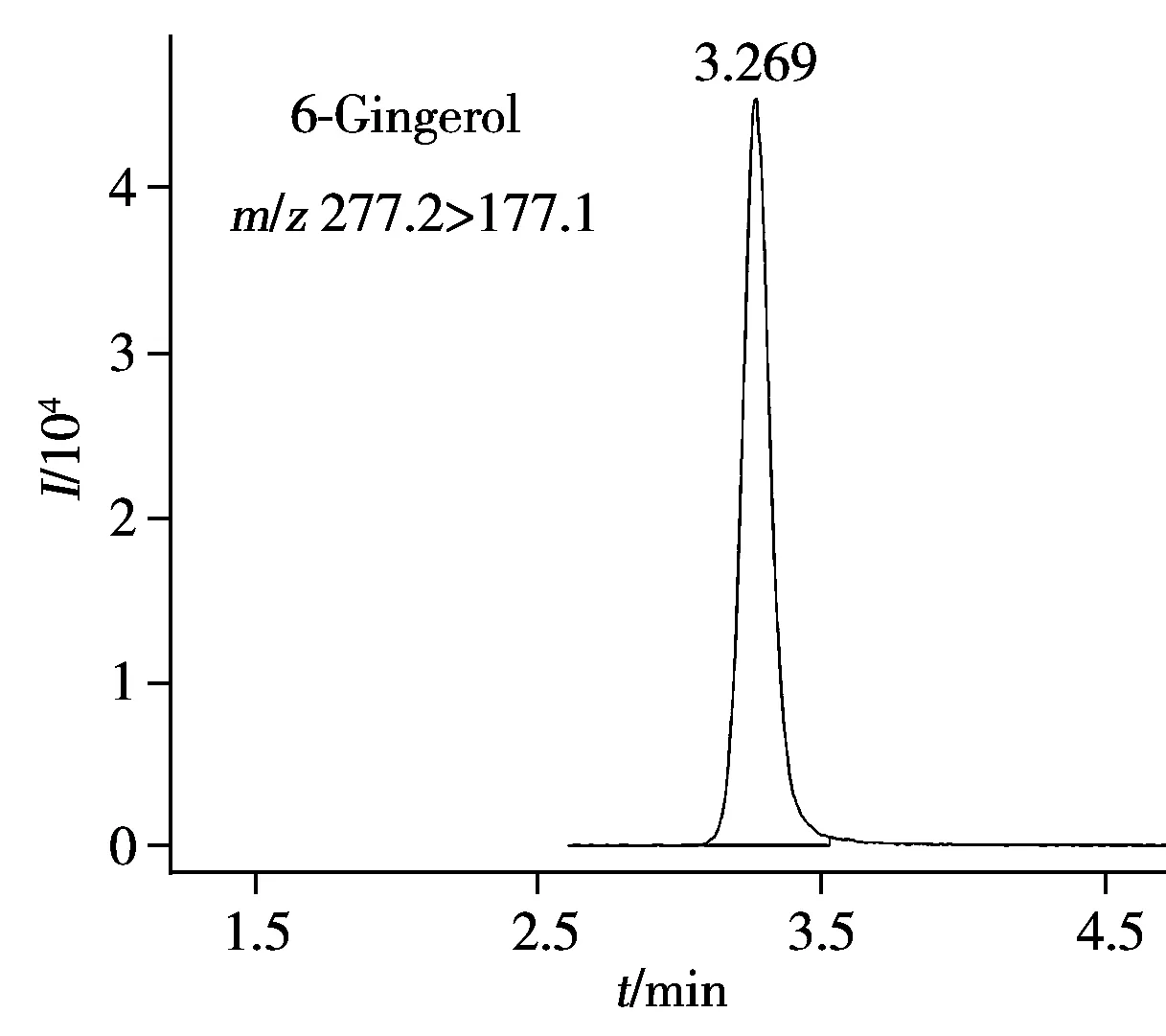
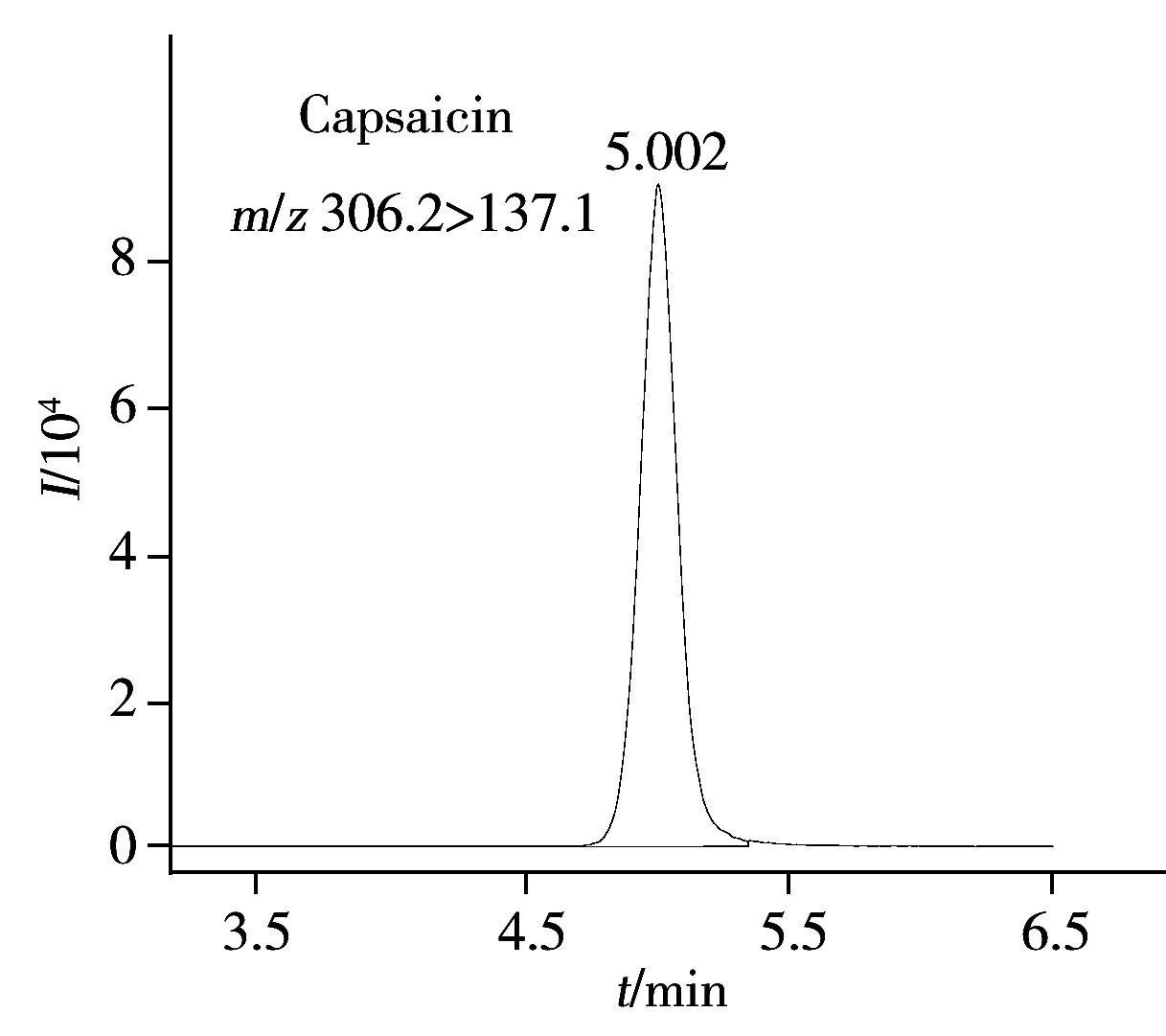
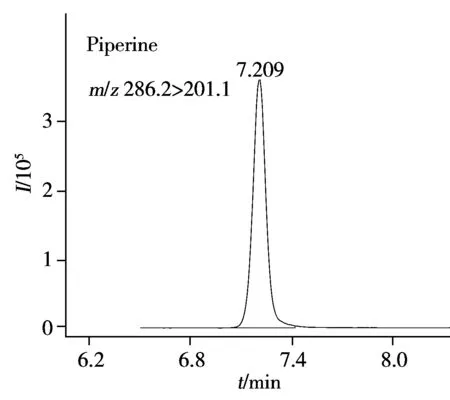
2.6 线性关系、检出限与定量下限
在优化的色谱-质谱条件下,对6个质量浓度水平的系列混合标准溶液(含25 μg/L内标物)进行测定。以各组分的MRM定量离子对峰面积与内标物定量离子对峰面积的比值为纵坐标(y),对应的质量浓度为横坐标(x,μg/L)绘制标准曲线,得到线性回归方程。由表2可知,各待测物均具有良好的线性关系,相关系数(r2)为0.996 5~0.999 7。采用标准添加法测定,以定量离子对的信噪比(S/N)不小于3确定待测物的检出限(LOD),S/N不小于10确定待测物的定量下限(LOQ),得到9种待测物的LOD为0.02~0.10 μg/kg,LOQ为0.05~0.25 μg/kg(见表2)。
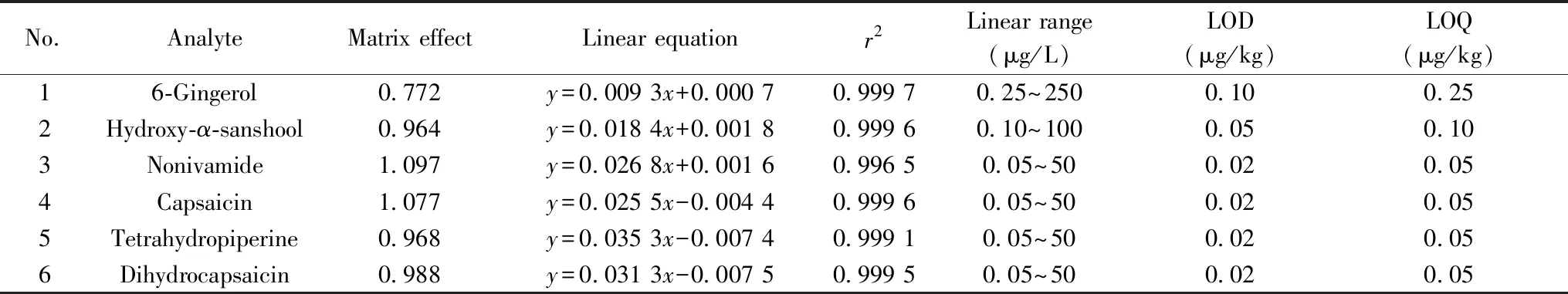
表2 9种待测物的基质效应、线性方程、相关系数、线性范围、检出限与定量下限Table 2 Matrix effects,linear equations,correlation coefficients(r2),linear ranges,LODs and LOQs of nine analytes
(续表2)
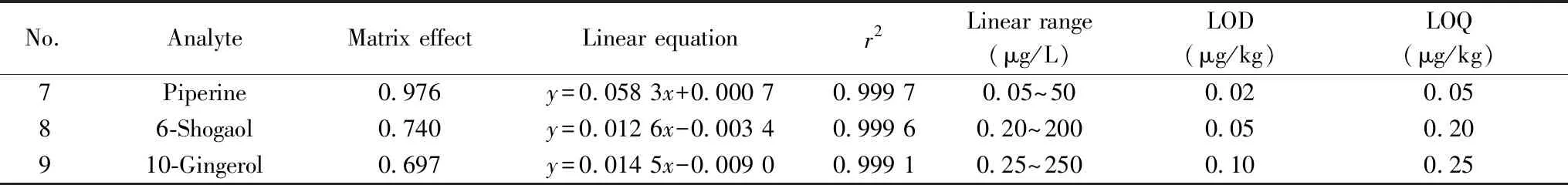
No.AnalyteMatrix effectLinear equationr2Linear range(μg/L)LOD(μg/kg)LOQ(μg/kg)7Piperine0.976y=0.058 3x+0.000 70.999 70.05~500.020.0586-Shogaol0.740y=0.012 6x-0.003 40.999 60.20~2000.050.20910-Gingerol0.697y=0.014 5x-0.009 00.999 10.25~2500.100.25
y:peak area ratio of the quantitative ions of analyte to the internal standard;x:mass concentration(μg/L)
2.7 回收率与相对标准偏差
取空白样品,在低、中、高3个添加水平下进行加标回收试验,每个加标水平按本法处理后测定,平行测定6次,计算待测物的平均回收率和相对标准偏差(RSD,n=6)。由表3可知,9种待测物的平均回收率为68.2%~104%,RSD为2.2%~12%。结果表明,方法具有较好的准确度和精密度。
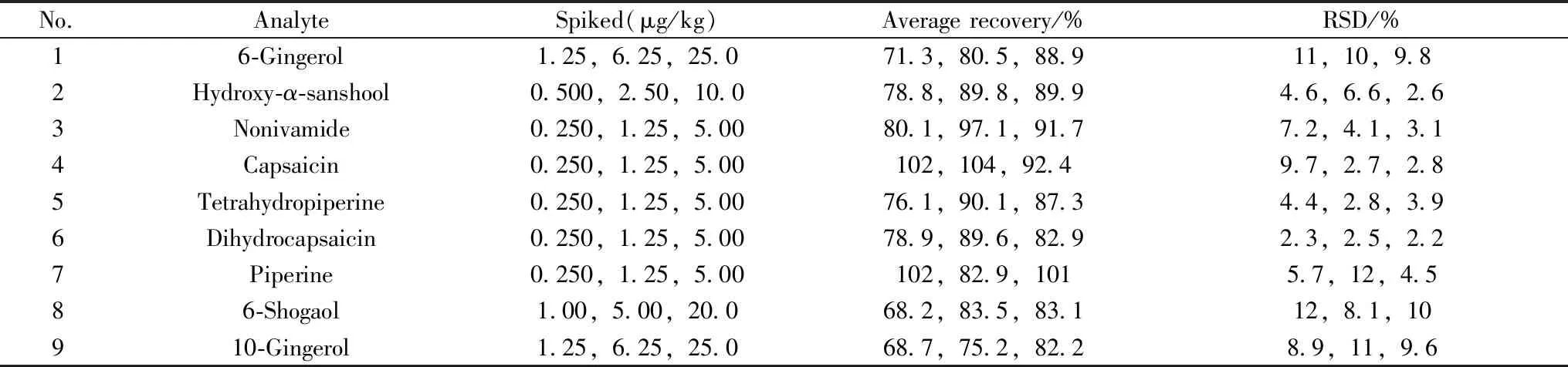
表3 9种待测物的平均回收率与相对标准偏差(n=6)Table 3 Average recoveries and relative standard deviations(RSD) of nine analytes(n=6)
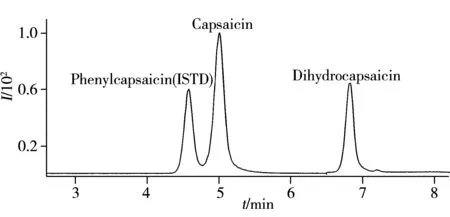
图3 阳性样品的总离子流(TIC)色谱图Fig.3 Total ion current(TIC) chromatogram of a positive sample
2.8 实际样品测定
应用本法筛查了购自本地区市郊某农贸市场的20批次食用油样品,其中1个批次样品检出较高含量的辣椒素(37.2 μg/kg)及二氢辣椒素(26.3 μg/kg),其余待测化合物均未检出,阳性样品的总离子流色谱图见图3。
3 结 论
本研究建立了UHPLC-MS/MS同时快速测定食用油中来源于4类调料的9种外源性特征杂质的分析方法。采用PFP色谱柱在10 min内实现了9种待测物的良好分离。与现有方法相比,该法样品前处理更为简便、高效,采用内标法校正,定量更加准确可靠,是同时测定食用油中难挥发外源性特征杂质种类最多的方法。该法适用于食用油的质量安全监测,为非正常来源食用油的鉴别提供了可靠方法,可为政府相关部门提高食用油监管水平提供科学依据和技术支持。
