足细胞损伤与糖尿病肾病
2019-05-22张馨元米焱综述王彩丽审校
张馨元 米焱 综述 王彩丽 审校
糖尿病肾病(DN)是糖尿病的主要微血管并发症之一,患病率约为20%~40%。若不及时干预,约80%的1型糖尿病和20%~40%的2型糖尿病伴微量白蛋白尿的患者在10~15年发展为临床肾病,最终进展为终末期肾病(ESRD)[1]。DN早期表现为微量白蛋白尿,因症状不明显,易被忽视,疾病逐渐演变可产生大量白蛋白尿伴肾功能下降,其发病机制较复杂,确切研究还未明确。最近的研究通过证明足细胞对维系肾小球滤过屏障的重要性,突出了足细胞及相关蛋白的损伤与DN之间的密切联系。
DN足细胞损伤机制
足细胞即肾小球脏层上皮细胞,其参与稳定肾小球毛细血管,维持肾小球滤过屏障的功能,调节超滤系数K/f以及保持肾小球基膜(GBM)的正常形态,是维持肾小球滤过屏障结构和功能正常的主要细胞之一[2]。研究表明,足细胞损伤在DN的发病机制中起着关键性作用[3],包括表观遗传因素、氧化应激、糖代谢异常、血流动力学改变、细胞因子、胰岛素抵抗等,这些变化导致各种细胞反应,分泌因子和细胞外基质的表达,最终导致肾小球滤过屏障的破坏,以及组织学变化,包括系膜扩张,结节性肾小球硬化和肾小管-间质纤维化。
表观遗传因素表观遗传修饰参与了DN等多种糖尿病并发症的发生发展。近日非编码RNA中的微小RNA(micro RNA,miRNA)在肾脏病理学中的作用受到广泛关注,特别是在DN方面。如miRNA-27a的敲低可减少链脲佐菌素(Streptozotocin,STZ)诱导的DN大鼠的肾脏细胞外基质(extracellular matrix,ECM)积累和尿蛋白[4]。此外,miRNA-27a/PPARr/β-连环蛋白轴能促进DN中足细胞损伤的进度[5]。研究发现,DN大鼠模型中增加的异黏蛋白(metadherin,MTDH)可通过下调miRNA-30s的表达而活化p38 MARK依赖通路促进足细胞凋亡[6]。组蛋白转录后修饰(post-translational histone modification,PTHMs)作为重要的表观遗传调控机制,和DN足细胞的损伤有着潜在的联系。其中,最具特点的为乙酰化和甲基化,这两种修饰均参加病理基因和保护基因的表达调控。Majumder等[7]特异性地从足细胞中删除组蛋白-赖氨酸N-甲基转移酶EZH2或者抑制EZH2活性系统从而降低H3K27me3标记的水平,引起Notch途径激活,进而促进足细胞去分化和加速肾小球损伤。另外,抑制赖氨酸特异性去甲基化酶,可通过改变组蛋白模式而有利地改善肾小球疾病的进展。甲基化、组蛋白修饰测序技术不仅仅是单纯的DNA测序,有望成为认识DN遗传易感性的一个新途径。
氧化应激DN时机体的氧化应激状态可通过多途径、多方面影响足细胞的形态和功能,造成足细胞损伤。高糖刺激晚期糖基化终产物(AGEs)的形成和积累,增加活性氧(ROS)的合成,从而引起持续的氧化应激。ROS主要由NADPH氧化酶介导的途径产生,过量的葡萄糖通过多种辅助途径代谢,如多元醇途径、蛋白激酶C(PKC)、肾素-血管紧张素-醛固酮系统(RAAS)等诱导足细胞凋亡。所有这些途径都是互为因果,相互关联的,即AGEs和PKC促进氧化应激,氧化应激反过来加剧了AGEs和PKC的产生[8]。进一步研究后发现足细胞中糖酵解及其代谢产物的改善可能诱导线粒体的再生,这可逆转表观遗传变化,或者加速细胞外基质更新和重塑,随后会改善肾小球功能,并延缓疾病的进展(图1)[9]。
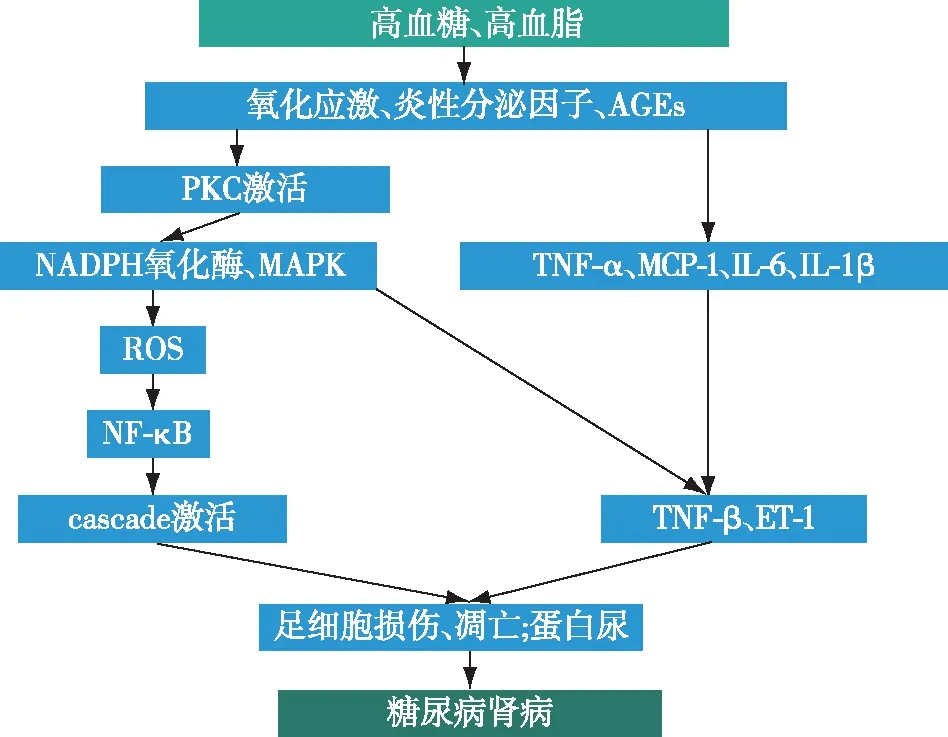
图1 氧化应激、糖代谢异常、炎性细胞因子等机制致糖尿病肾病示意图糖尿病肾病时,糖、脂代谢异常、氧化应激、炎性细胞因子等多种途径致足细胞损伤、凋亡,并出现蛋白尿;AGEs:晚期糖基化终产物;PKC:蛋白激酶C;TNF-α:肿瘤坏死因子α;MCP-1:人单核细胞趋化蛋白1;IL-6:白细胞介素6;IL-1β:白细胞介素1β;NADPH:烟酰胺腺嘌呤二核苷酸磷酸;MAPK:丝裂原活化蛋白激酶通路;ROS:活性氧;NF-κB:核因子κB;caspase:含半胱氨酸的天冬氨酸蛋白水解酶;TGF-β:转化生长因子β;ET-1:内皮素1
糖代谢异常高糖直接增加肾小球系膜细胞的氧化应激,包括葡萄糖自氧化和高血糖引起的代谢应激。高糖状态下葡萄糖与游离氨基酸或组织蛋白结合,并最终形成不可逆AGEs,而后者可直接引起足细胞损伤[10]。生理条件下肾脏晚期糖基化终末化产物受体(RAGE)主要在足细胞表达,DN时足细胞RAGE表达增加,因而推测AGEs可能是通过RAGE介导的机制损伤足细胞。AGE与细胞膜上的RAGE结合后,激活多条信号转导通路等,增加炎症因子等的表达,最终改变组织细胞的功能,甚至产生组织破坏,从而促进DN进展[11]。有研究发现AGEs通过氧化应激反应可引起胰岛β细胞合成及释放胰岛素功能障碍,诱导β细胞凋亡,还可抑制胰岛β细胞发生自噬作用从而促进胰岛β细胞凋亡[12]。
血流动力学改变DN早期即可出现血流动力学异常,高血糖、AGE及高血压等机械应力都会使血管紧张素Ⅱ(Ang Ⅱ)水平增加。AngⅡ通过改变足细胞蛋白表达及分布对足细胞造成直接损伤。此外,AngⅡ还促使细胞增生、增加细胞凋亡[13]。Ang Ⅱ诱导的氧化应激可激活足细胞中处于潜伏期的转化生长因子β (TGF-β)活性增强,进而导致GBM增厚和足细胞损伤等。TGF-β也可能导致足细胞凋亡和启动时,与上皮间质转化(EMT)一起脱离GBM造成肾小球的硬化。肾小球毛细血管张力及压力增高使AngⅡ、生长因子等合成和释放增多,从而促进DN的发展[14]。研究发现,血管内皮生长因子(VEGF)及其受体在DN时上调,与肾小球高滤过、蛋白尿、肾小球肥大有关[15]。DN时足细胞和血清中的VEGF水平增强,表明VEGF可能有助于DN的发病。有研究报道增强自噬可以抑制VEGF的表达,对足细胞功能的维持十分重要[16]。
炎性细胞因子炎性细胞因子如巨噬细胞集落刺激因子、肿瘤坏死因子(TNF-α)、白细胞介素6 (IL-6) 、IL-1β、核因子κB (NF-κB) 等均参与了DN的发生发展[17]。研究中,可以发现糖尿病大鼠的肾脏与血清中TGF-β和IL-10的增加,糖尿病中AGE介导的ROS引起促进TGF-β和结缔组织等硬化炎性因子的产生。一些研究证实了NF-κB介导的炎症通路诱发了糖尿病动物ECM积聚和足细胞产生损伤[18]。另外,蛋白S是炎症反应的负调节因子,和TNF-α诱导的NF-κB信号传导是DN发病机制的驱动因素。培养的足细胞中的PS过表达抑制了高葡萄糖和TNF-α诱导的促炎反应[19]。上述炎症因子与氧化应激、糖代谢紊乱等相互作用从而引发一系列的损伤。
胰岛素抵抗胰岛素抵抗(IR)是指靶细胞对胰岛素的敏感性降低或胰岛素介导的信号传导障碍。研究表明足细胞为胰岛素敏感细胞[20],炎性因子与脂代谢紊乱等因素均参与了胰岛素抵抗。Reidy 等[21]研究显示糖尿病早期足细胞就已经出现了特异性的胰岛素抵抗,再次说明了破坏足细胞上正常的胰岛素信号传导是DN的发生发展的病理分子机制。
足细胞相关蛋白与DN的相关性
足细胞相关蛋白按其功能区域可分为基膜区蛋白、顶膜区蛋白、裂孔隔膜(SD)蛋白及细胞骨架相关蛋白,足细胞相关蛋白表达异常,参与或导致了DN的进程。
基膜区蛋白的异常足细胞基膜区表达一系列的细胞-基质黏附受体,整合素α3β1是一种重要的受体,可以将足细胞与GBM紧密连接,还参与细胞骨架的形成[22]。然而,在病理条件下,整合素α3β1会使足细胞的迁移或脱落增加,并且在DN的早期阶段就可以观察到整合素水平的变化[23],高血糖可以下调人和大鼠整合素α3β1的表达,并触发整合素连接激酶(ILK)的激活。 ILK是足细胞生物学必不可少的。 ILK的异常影响足细胞的黏附能力从而诱导足细胞与GBM分离。有研究表明AGEs可直接抑制足细胞的黏附能力,上调ILK表达和激活局部RAS[24]。此外,高血糖诱导的ROS也会降低α3β1整合素表达并最终导致足细胞脱离GBM。最近的研究表明细胞内整合素β1和整合素β3表达增加,足细胞运动能力增强。这可能是其在HG条件下对足细胞损伤具有保护作用的潜在机制之一[25]。
顶膜区蛋白的异常足细胞表面标志蛋白podocalyxin(PCX)是足突顶膜区主要的带负电荷的唾液蛋白,对保持肾小球滤过功能的完整性有着重要作用。事实上,SD相关蛋白的损伤和mRNA的表达与其PCX尿、蛋白尿程度有关,尿PXC水平越高则肾小球的损害越严重,进而可以作为预测DN进程的生物标志物[26]。另外,Economou等[27]在STZ诱导的糖尿病大鼠模型中发现,PCX的表达较正常对照组下降45%,以高糖刺激体外培养足细胞后PCX的表达几乎全部被抑制,故认为DN时podocalyxin表达量下降,肾小球滤过电荷屏障减弱,促进蛋白尿的发生,加速DN的进展。
SD蛋白的异常裂孔膜蛋白分子包括nephrin、podocin、CD2AP、NEPH1、densin、P-cadherin、ZO-1、FAT等,其中较关键的SD分子有nephrin、podocin、CD2AP、TRPC6,它们参与稳定SD的完整性。Nephrin作为SD的核心,也是足细胞中的信号传导中枢,调节细胞极性、黏附,维持细胞骨架,参与足细胞生存等。nephrin在SD中的表达对于屏障维持至关重要,其通过podocin和CD2AP与肌动蛋白细胞骨架相连,其中任何一种蛋白质的突变都可能导致足细胞损伤和大量蛋白尿。已有研究发现,Nephrin基因的启动子区存在DNA甲基化和组蛋白修饰调控,并且其表遗传修饰水平受到转录因子Kruppel样因子4 (kruppel-like factor4,KLF4) 的调节。KLF4 是一种具有锌指结构的转录因子,在肾脏发育及分化时期发挥重要的转录调控作用,与足细胞标志基因表达密切相关[28]。podocin 是继 nephrin 后发现的另一种特异性表达于SD的相关蛋白。研究发现 DN 患者足细胞 podocin 蛋白表达降低,且其表达减少与尿蛋白增加相关[29]。nephrin、podocin 和neph1组成 nephrin-podocin-neph1 受体复合物,编码这些蛋白的基因缺失及突变,将导致肾小球滤过功能障碍,该复合物以及其他细胞间连接蛋白与细胞骨架相关蛋白相互作用,从而调节足突细胞骨架的形态学和动力。CD2AP 是维持 SD 复合体结构及功能的另一种重要蛋白,除直接与 podocin 等多种 SD 蛋白发生作用外,同时还可作为桥接蛋白与细胞骨架蛋白[α辅机动蛋白4(α-actinin-4)] 发生联系以保持足突处于正常状态[30]。CD2AP 与 podocin 在足细胞内 SD 处发生连接,正常情况下它们在足突和SD的形成中发挥着协同作用,CD2AP 蛋白表达的减少,可同时伴 podocin蛋白表达降低,它们的减少导致了足细胞发育停滞,与蛋白尿的发生与发展、肾小球的病变有着密切关系。TRPC6与nephrin在足细胞存在共定位分布,并且二者之间存在直接联系。TRPC6高表达能减少足细胞nephrin的表达,但对其分布无明显影响,这可能是TRPC6参与足细胞损伤、加重蛋白尿的机制之一[31]。研究表明Ang Ⅱ可通过刺激炎症介质分泌而激活足细胞中的TRPC6通道加重白蛋白尿[32](图2)。

图2 糖尿病肾病中足细胞相关蛋白异常表达高血糖、TGF-β/Smad等多种途径、通路影响足细胞正常形态从而使足细胞相关蛋白表达异常进而加重糖尿病肾病的发展;EMT:上皮细胞-间充质转化;TRPC6:瞬时受体电位阳离子通道6;P-cad:钙黏蛋白;ZO-1:紧密连接蛋白;TGF-β:;转化生长因子β;CD2AP:CD2相关蛋白;Densin:密度蛋白;NEPH1:肾病样蛋白1抗体
最近发现在人和小鼠足细胞中特异性表达的srGAP2a蛋白,其与足细胞标志蛋白synaptopodin共定位,DN患者和db/db小鼠肾小球中表达显著下降。srGAP2a蛋白与DN临床表型蛋白尿和eGFR密切相关,对维持足细胞细胞骨架的稳定性起着重要作用,其主要是通过抑制RhoA和Cdc42的过度活化来稳定F-actin。在DN状态下,srGAP2a表达显著下调,可能促使RhoA和Cdc42活性增加,从而出现足细胞迁移增加,骨架重排,进而足细胞从基底膜上脱落。外源性给予srGAP2a可缓解DN的足细胞损伤和蛋白尿的产生[33]。
细胞骨架蛋白的异常细胞骨架是维持足细胞结构和功能的核心因素,对于足细胞细胞骨架的持续性调节是维持肾小球滤过屏障的至关重要的因素[34]。目前已发现的足细胞骨架蛋白有α-actinin-4、synaptopodin、波形蛋白、结蛋白(desmin)和巢蛋白(Nestin)等。细胞骨架蛋白最主要的分子组成是F-actin,其相关蛋白α-actinin-4、synaptopodin等将F-actin交联在一起,起桥梁作用,分别连接基膜的整合素和裂孔膜蛋白,对维持足细胞正常形态功能起重要作用。
小结:DN时,足细胞损伤机制及相关蛋白表达的改变,将导致肾小球滤过屏障结构和功能的异常,从而促使尿蛋白形成和或肾小球硬化。因此,如果能有效地改变 DN 时足细胞结构或功能或改变其相关分子的表达状况,有望在 DN 的防治上取得突破性进展。
