毛细血管渗漏综合征研究进展
2019-05-22董建华综述李世军审校
董建华 综述 李世军 审校
毛细血管渗漏综合征(capillary leak syndrome,CLS)是系统性炎性反应综合征的一种严重并发症,以体液和蛋白从血管渗漏到组织间隙为特征,临床表现为全身进行性水肿、胸腔和腹腔积液、少尿、低血压、低氧血症和低白蛋白血症,可累及全身多个脏器,尤其是肺和肾脏[1]。 脓毒症等多种疾病可是诱发CLS。本文拟对CLS的病因、病理生理机制、临床表现和治疗方法进行综述。
病 因
CLS的常见病因有感染、创伤、烧伤、急性重症胰腺炎、体外循环术后等,脓毒症最常见。内毒素刺激炎症细胞释放大量细胞因子,如肿瘤坏死因子α (TNF-α),白细胞介素1(IL-1)、IL-2、IL-6等,进一步激活多形核白细胞和内皮细胞等效应细胞,刺激氧自由基等释放,加速花生四烯酸代谢并释放血栓素A2、前列腺素等炎性介质[2]。在炎性介质作用下,毛细血管内皮细胞损伤、收缩,细胞连接分离,血管通透性增加,白蛋白等大分子物质渗漏到组织间隙,组织间隙胶体渗透压升高,血管内水分进入组织间隙,进而引起全身水肿和有效循环血量减少,导致器官缺血、缺氧,临床可表现为低血容量休克、低蛋白血症、组织水肿。
系统性毛细血管渗漏综合征(systemic capillary leak syndrome,SCLS)是一种罕见疾病,也称Clarkson病,其特征是血管渗透性间歇性增加,表现为低容量性低血压、血液浓缩、横纹肌溶解、低蛋白血症、血单克隆免疫球蛋白、全身水肿、急性肾损伤(AKI)等临床症状反复发作,急性发作与病情缓解常交替出现,SCLS病因尚不明确[3]。梅奥医学中心回顾既往26年临床病例,仅25例诊断为SCLS[4]。个案报道自身免疫性疾病可以合并SCLS,如抗磷脂抗体综合征[5]、干燥综合征、系统性硬化症和多肌炎[6]。
肾综合征出血热(hemorrhagic fever with renal syndrome,HFRS)是由汉坦病毒引起的自然疫源性疾病,以发热、出血、血小板减少、低血压休克和肾脏损害为主要临床表现[7]。典型病程经过发热期、低血压休克期、少尿期、多尿期及恢复期。AKI在HFRS中常见,可出现蛋白尿和血尿[8],肾脏病理常表现为急性间质性肾炎[9]。
噬血细胞性淋巴组织细胞增生症(hemophagocytic lymphohistiocytosis,HLH),又称噬血细胞综合征,是由于细胞毒T淋巴细胞和自然杀伤细胞的细胞毒效应显著降低和功能障碍,不能及时有效清除病毒等抗原,巨噬细胞持续异常活化和增生所致的多器官高炎症反应和组织器官免疫损伤的临床综合征[10]。大量淋巴细胞和巨噬细胞活化会导致高细胞因子血症。约16%~62%的HLH患者出现AKI,常有肾前性诱因,可也合并急性肾小管坏死(acute tubular necrosis,ATN)、血栓性微血管病导致AKI[11]。继发于感染的HLH可应用抗生素和丙种球蛋白,继发于自身免疫性疾病、恶性肿瘤的HLH均可使用糖皮质激素治疗[12]。
植入综合征发生于造血干细胞移植后中性粒细胞恢复过程中,以发热、低血压、水肿、胸腔及腹腔积液、非心源性肺水肿和AKI为主要表现的临床综合征[13-14],可采用激素治疗[15]。
卵巢过度刺激综合征(ovarian hyperstimulation syndrome,OHSS)是体外受孕辅助生育的主要并发症,是人体对促排卵药物产生的过度反应。外源性人绒毛膜促性腺激素会促进多个卵泡发育,释放大量血管内皮生长因子(vascular endothelial growth factor,VEGF),腹腔内VEGF浓度明显高于血清。约2%OHSS患者出现卵巢增大、大量腹腔积液,引起呼吸困难和AKI[16]。Abramov等[17]分析209例重度OHSS患者,29.7%出现少尿,1.4%发生AKI。
分化综合征是急性早幼粒细胞白血病患者在接受全反式维甲酸或三氧化二砷诱导治疗过程中出现的并发症。分化的急性早幼粒细胞释放大量细胞因子,白血病细胞和靶组织的整合素和黏附分子表达增加,急性早幼粒细胞会浸润靶器官,特别是肺。通常发生在治疗开始后10~12d,特征性表现为发热、体重增加、急性呼吸窘迫综合征(acute respiratory distress syndrome,ARDS)、胸腔积液、全身水肿、低血压和AKI,AKI的发生率为28%。激素可有效治疗分化综合征引起的CLS[18]。
部分抗肿瘤药物可引起CLS。吉西他滨治疗恶性肿瘤后IL-2和TNF-α显著升高,导致全身水肿、低血压、非心源性肺水肿和AKI[19]。单克隆抗体能够快速激活大量免疫细胞,导致多种细胞因子的骤然增加,一般在用药数分钟至数小时后出现症状。利妥昔单抗治疗慢性淋巴细胞白血病时,会引起TNF-a和IL-6增加,当淋巴细胞计数>50×109/L,发生率严重CLS的风险高[20]。激素能有效改善药物引起的CLS。
病理生理机制
CLS的根本原因是高细胞因子血症,导致毛细血管对蛋白质的通透性增高,富含蛋白质的液体通过毛细血管屏障进入组织间隙。分子量200~900 KD的血浆物质均可渗漏到组织间隙。白蛋白分子量66.5 KD,在CLS的前12h,血白蛋白渗出30%~50%。内皮细胞保持血管通透性的能力取决于相邻内皮细胞间黏着连接和紧密连接的完整性,并以黏着连接的作用最为重要。血管内皮钙粘蛋白(vascular endothelial cadherin,VECD)是黏着连接的主要成分。轻度炎症刺激引起VECD内化,血管通透性增加,但内皮结构保持完整。严重炎症反应导致内皮细胞间隙和渗透性显著增加[21]。脓毒症可破坏黏着连接增加血管通透性。用CLS急性发作期患者的血清孵育微血管内皮细胞,VECD减少,黏着连接的完整性降低,微血管通透性增加。而用静止期患者血清孵育血管内皮细胞,黏着连接无明显变化。血浆中的可溶性因子会增加血管内皮渗透性[22]。VEGF是已知增加毛细血管通透性作用最强的物质,可使VECD内移。毛细血管通透性增加是脓毒症发生发展的重要因素,VEGF是控制毛细血管通透性的关键分子,是脓毒症诱发CLS的潜在因素[23]。虽然还没有研究证实其他病因引起的CLS中内皮细胞连接的改变,但是这些疾病共同的临床表现,提示其存在共同的病理生理机制。
临床表现
CLS临床可分为渗漏期和恢复期两个阶段[24]。渗漏期表现为进行性全身水肿、胸腔及腹腔积液、胃肠道水肿、非心源性肺水肿、低蛋白血症、低血压、少尿等,若治疗不及时,可因组织灌注不足而发生多器官功能障碍综合征。恢复期表现为全身水肿逐渐消退、血压及中心静脉压回升、不使用利尿剂的情况下尿量自行增加等,易发生非心源性肺水肿。
血流动力学紊乱富含蛋白质的液体从血管渗漏至组织间隙,有效循环血量不足,引起肾素、血管紧张素、醛固酮系统和交感神经系统激活及血管加压素释放,而水钠潴留会导致全身水肿和渗出性浆膜腔积液[17]。CLS急性发作时,如SCLS、HFRS、OHSS等,均可伴有血液浓缩和低血容量休克(图1)。血液浓缩可作为毛细血管渗漏严重程度的标志。
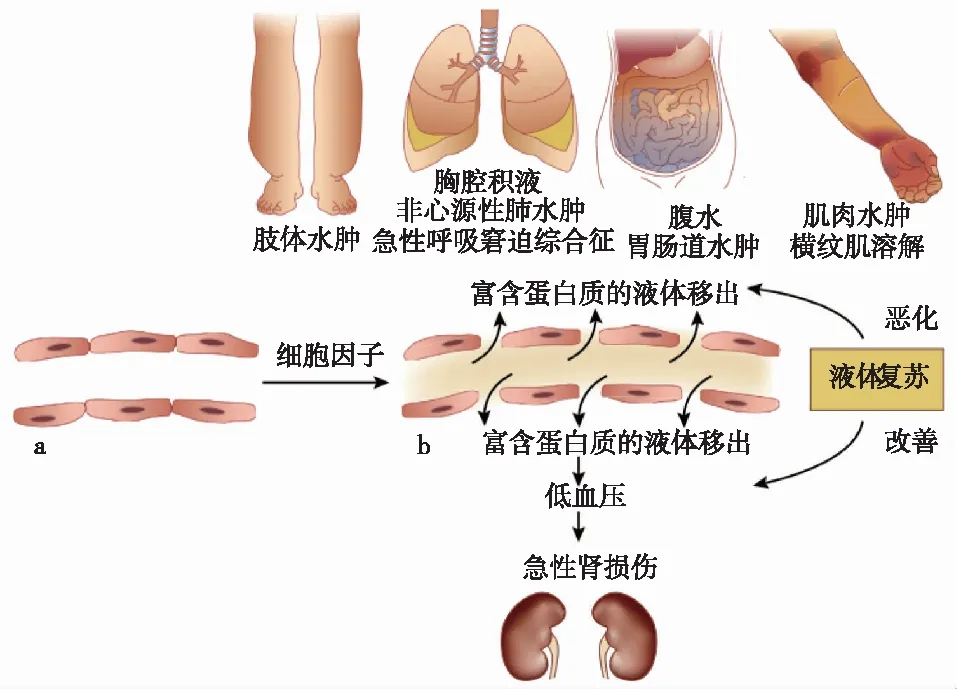
图1 毛细血管渗漏综合征的临床表现
肾脏损伤以AKI是常见,有效循环血量不足所致的肾前性因素是引起AKI最常见的原因,其次是ATN[4]。细胞因子在ATN的发生中可能有重要作用。某些SCLS病例可因横纹肌溶解引发的肌红蛋白尿而导致ATN[4]。少数HLH患者会出现肾病综合征[11]。HFRS患者早期可能出现短暂的蛋白尿和(或)血尿,提示存在轻度肾小球损伤,与肾前性因素和ATN叠加[8,9]。OHSS因张力性腹水引起腹腔间隔综合征,导致AKI[17]。
肺损伤CLS均可发生胸腔积液。CLS渗漏期易出现非心源性肺水肿。HLH和分化综合征可出现肺出血,而SCLS、HLH、OHSS、植入综合征和药物诱导的CLS可出现ARDS。呼吸困难是OHSS常见的呼吸系统症状,主要是因为大量腹水限制横膈膜下移,肺容量降低[17]。
其他目前尚缺乏CLS的其他器官系统受累数据。SCLS患者可出现肌肉水肿所致的横纹肌溶解和筋膜室综合征[4]。恶心、呕吐和腹痛在CLS中很常见。
诊 断
临床尚未制定CLS诊断标准。Marx等[25]通过吲哚菁绿测量血浆容量、铬51标记红细胞测量红细胞容积、菊粉测量细胞外液,并观察胶体渗透压的改变,发现CLS患者较非CLS细胞外液明显增加,而胶体渗透压升高不明显;该方法价格昂贵、操作复杂,临床应用有限。目前诊断CLS主要依据存在诱发因素,同时出现中心静脉压下降(<5 cmH2O),血清白蛋白明显降低(<25 g/L),氧合指数降低(<300 mmHg);胸部影像学提示肺间质渗出性改变;血细胞比容增加;全身进行性水肿,伴有腹腔、胸腔积液、体重增加等临床表现。CLS可能被归因于低白蛋白血症,故需排除肾源性、心源性、肝源性、神经源性及其他不明原因所致的水肿。遗传性血管性水肿、Gleich综合征(发作性血管性水肿伴嗜酸性粒细胞增多)可伴有皮肤、呼吸道和胃肠道水肿,容易与CLS混淆。 临床上还需与以循环衰竭为主的过敏性休克、中毒性休克相鉴别。目前可利用脉波轮廓温度稀释连续心排血量监测技术(PiCCO)监测肺血管通透性,从而了解周身毛细血管通透性状况,为CLS的诊断和治疗提供更客观数据支持。
治 疗
CLS尚无特异性治疗方法,临床多在治疗原发病基础上,维持有效循环血量,改善毛细血管通透性,预防重要脏器缺血缺氧。
液体治疗是CLS治疗的重要措施(图2)。根据不同阶段的病理生理特点选择恰当的液体治疗是维持组织器官的灌注并减少并发症的关键。临床常用的液体包括晶体液和胶体液。晶体液分布容积大,输入后可迅速进入细胞外液,既补充血容量也补充组织间质容量的缺失,缺点是半衰期短,往往需要补充失液量的4~6倍且重复使用才能维持血容量。大量的晶体液可稀释血浆白蛋白,降低血浆胶体渗透压,输入后很快扩散至组织间,导致组织水肿加重,氧的弥散距离增大,进一步加重组织缺氧。CLS的渗漏期,毛细血管通透性明显增大,晶体液更容易渗漏到组织间,血容量难以维持,因此一般不作为首选。

图2 毛细血管渗漏综合征的治疗
胶体液包括白蛋白、血浆等天然胶体和高分子量淀粉等人工胶体液。白蛋白占血浆胶体渗透浓度的80%。CLS时输注白蛋白仅可短时间内增加血容量,随后白蛋白渗漏到组织间隙,使组织间隙胶体渗透浓度增高,导致更多的水分积聚在组织间,维持血容量的效果欠佳,因此CLS扩容时要少用白蛋白。血浆主要用于纠正凝血功能障碍,但不作为常规扩容剂使用。高分子量淀粉如喷他淀粉(分子量264 kD)和羟乙基淀粉(分子量450 kD)不易透过血管内皮和渗漏至组织间隙,可有效增加血容量,且维持时间长,理论上可以作为CLS的复苏液体。一项病例报告示2例SCLS患者在应用喷他淀粉后有效提高中心静脉压和血压,其中1例应用9L生理盐水扩容后,血压、中心静脉压仍低,予喷他淀粉治疗后,血流动力学逐渐稳定[26]。另一项病例报告描述了1例SCLS患者接受大量晶体液复苏后导致下肢筋膜室综合征,而随后的多次复发均予羟乙基淀粉联合晶体液治疗,未再发生液体过负荷并发症[27]。比较重症患者使用羟乙基淀粉和其他复苏液(包括晶体液、白蛋白和凝胶等)进行液体复苏的情况,发现羟乙基淀粉不能降低患者死亡率,而且显著增加患者死亡率和AKI发生率,并增加肾脏替代治疗的概率[28]。但是,CLS患者大多死于休克期,所以对于难治性休克患者,羟乙基淀粉的收益大于风险。足够容量复苏后仍处于低血压状态的患者,可应用血管活性药物提升血压。
在CLS恢复期患者易发生非心源性肺水肿,为避免大量液体回渗,建议使用液体限制策略,需重新评估血管内容量、肺水状态及组织灌注情况。血压持续稳定,应早期应用利尿剂,防止肺水肿。容量过负荷者,若血流动力学稳定,首选袢利尿剂减轻容量负荷;若血压偏低,器官组织灌注尚可,可应用白蛋白联合利尿剂减轻容量负荷。利尿剂反应差伴AKI的患者需行肾脏替代治疗。
连续性肾脏替代治疗(CRRT)不仅可维持机体内环境稳定,也可清除细胞因子、调节机体免疫功能。CRRT可清除炎症介质,减轻血管内皮细胞损伤,改善毛细血管渗漏,防止病情进展,提高存活率。CRRT同时能缓慢清除体内多余体液,降低组织和器官水肿,又能补充足有效循环血量,降低低血压发生率,维持机体血流动力学稳定。血流动力学不稳定者,CRRT上机前需要液体预充透析管路,防止血液进入管路后血压下降,采用血白蛋白预充管路,对维持血浆胶体渗透压的效果好、安全性高。重度OHSS出现大量腹水时,腹腔穿刺引流可减轻压迫症状;出现ARDS时,应予呼吸机辅助呼吸,维持有效通气和换气功能,改善氧合,纠正低氧血症。
糖皮质激素具有抑制炎症反应、降低毛细血管通透性的作用,对药物、植入综合征、分化综合征、HLH和自身免疫性疾病引起的CLS均有效。部分CLS对激素无效,可能是因为导致内皮细胞损伤的因素不受激素影响。免疫球蛋白(Ig,分子量146 kD)可能是目前治疗CLS和预防复发的最有效方案,但临床证据有限,数据多来源于感染继发性HLH。Ig在CLS急性期可透过血管内皮,渗漏至组织间隙,不能提供持续的胶体渗透压。Ig对CLS有效,可能与其本身免疫调节作用有关[29]:(1) 阻断白细胞表面Fc受体,控制炎症反应;(2)具有抗细胞因子特性,抑制白细胞介素产生,尤其是IL-2。目前报道疑似CLS患者应立即静脉注射Ig,剂量为1 g/(kg·d)×2d[29],预防剂量为2 g/(kg·月)[30]。
小结:脓毒症、SCLS、HFRS、HLH、植入综合征、OHSS、分化综合征等多种疾病及抗肿瘤药物均可诱发CLS。毛细血管通透性增高,大量富含蛋白质的液体渗出是其共同的病理生理机制。高细胞因子血症是CLS的根本原因。CLS常引起AKI。液体治疗是治疗CLS的关键环节,人工胶体可作为CLS渗漏期的复苏液体,恢复期易发生非心源性肺水肿。糖皮质激素对多种疾病诱发的CLS有效。大剂量免疫球蛋白可能是治疗CLS的有效方法。
