一种面向大规模序列数据的交互特征并行挖掘算法
2019-05-15赵宇海汪嗣尧王国仁
赵宇海 印 莹 李 源 汪嗣尧 王国仁
(东北大学计算机科学与工程学院 沈阳 110819)
序列数据是一种重要的数据类型,被广泛应用于科学与工程学、商业、客户行为分析、股票趋势预测、DNA序列分析、Web使用行为分析等诸多实际应用领域[1-2].大数据时代,信息技术的不断廉价化与互联网及其延伸所带来的无处不在的信息技术应用,进一步加剧了海量序列数据的积累.以生物领域为例,高通量测序技术,尤其是新一代测序技术的迅猛发展,生物序列数据的规模呈爆炸性增长.大规模生物序列分析成为当前生物信息领域的热点研究问题之一[3-5].
基于单核苷酸多态性(single nucleotide polymor-phism, SNP)数据的复杂疾病全基因组关联研究是大规模生物序列分析的典型应用案例之一,其目的是在基因组范围内以SNP为标记,通过患病人群和正常人群之间的对比,找出显著序列变异,然后从中筛选出与疾病相关的位点(locus).该问题本质上可建模为特征选择问题,即从数据中选择出与分类属性关联性最强的特征子集.虽然问题简单明了,但解决起来却并不轻松,面临着一些严峻的挑战.当前发现的致病SNP中只有少数能中等或大量地增加复杂疾病的致病风险,一些已经被生物实验证实的致病SNP位点locus没有被GWAS(genome-wide associa-tion study)识别出来.研究结果仅解释了疾病很小部分的遗传变异,出现了“遗传丢失”现象,甚至彼此矛盾[3,6].例如,目前已发现了超过40个位点的变异影响人类身高性状,但在几万个样本实验中发现这些位点大概仅解释了5%左右的表型变异[3];类似地,在糖尿病研究中发现的18个位点仅解释了6%左右的遗传风险;再以儿茶酚氧位甲基转移酶基因COMT(catechol-O-methyltransferase)为例,Kotler等人[7]的研究支持158A(Met)会升高精神分裂症的发病率,而Wonodi等人[8]的研究则支持它会降低发病率.
造成以上情况的主要原因之一在于,传统GWAS研究仅仅关注了单点关联分析,忽略了微效位点间的交互作用对疾病的影响.生物体是个复杂系统,内部元件之间存在着错综复杂的关系,研究位点间的交互作用是解析复杂疾病性状遗传机理的重要途径之一.很多遗传学家相信位点间的交互作用在复杂疾病性状的遗传变异中扮演着重要角色.越来越多的理论和实验证据表明:SNP交互作用是复杂疾病产生和发展的重要遗传学基础之一[3-5].研究者们已在对老年痴呆症、乳腺癌、精神分裂症等复杂疾病的研究中发现了真实的SNP交互作用.全基因组SNP交互作用研究为GWAS开辟了崭新的研究思路和科研方向[1,5,9].针对复杂疾病,开展基于交互作用的高通量生物数据挖掘与分析技术研究成为新一波生物信息研究浪潮中人们关注的焦点[1,5,9].
SNP交互作用发现面临着严峻的计算挑战.全基因组关联研究中的SNP数目动辄几十万到上百万.即使只检验2位点互作,在30万个位点上的检测次数也已高达1 000亿次,对3阶甚至更高阶的交互作用更是难以想象的天文数字.例如假设每秒可以检测105个SNP位点组合,则仅3阶交互作用计算就需要约1.4×108年.因此,虽然目前生物上已证实高阶交互作用在复杂疾病中真实存在[10-11],但鲜有涉及该领域的研究报道.
针对大规模生物序列中高阶交互特征挖掘面临的密集计算问题,本文提出了一种新的基于并行处理和演化计算的解决框架,大致由4个主要步骤组成:
1) 数据预处理. 提出一种基于块和位点的2阶段过滤方法,提前过滤无关特征,提高整个框架的执行效率.
2) “低耦合高内聚”的数据划分. 执行序列挖掘任务,将原始高维特征数据划分成一系列低维特征区域,并保证其相互间的低交互可能性(即每个区域内的特征交互可能性高,不同区域间的特征交互可能性低).
3) 多样性的特征区域筛选. 许多划分后的特征区域具有较高相似度,如果在所有特征区域上都执行交互特征挖掘,会产生很多重复结果,降低计算效率. 将采用最小独立支配集的思想,基于多样性原则进行代表性特征区域选择.
4) 交互特征挖掘.提出基于置换搜索的并行蚁群算法,进行高效且有效的交互特征选择.
本文的主要贡献有4个方面:
1) 提出了一种面向大规模序列数据的交互特征并行挖掘框架.现有框架大多从减少各节点处理的序列数角度提高计算效率,因此对数据进行水平分组.与此不同,提出的框架从减少分组数据位点数入手,对数据进行垂直分解.位点数是制约互作挖掘效率的根本因素,提出的框架具有天然的优势.
2) 提出了极大等位公共子序列(maximal allelic common subsequence, MACS)的概念并设计了基于MACS的特征区域划分策略.该策略能保证交互特征在划分区域间的低耦合性和划分区域内的高内聚性,避免计算中的耦合引起的高额通信代价.
3) 设计并实现了一种基于置换策略的并行蚁群局部搜索算法(parallel ant colony optimization for interactive features search, PACOIFS),避免了特征子集搜索过程中的边际作用影响并保持计算的高效性.
4) 通过将本文算法和其他3个算法在糖尿病患者数据集和模拟数据集上进行测试分析,证明了算法的高效性和生物有效性.
1 相关工作
根据搜索策略,目前的交互特征挖掘方法可分为:枚举搜索、随机搜索和贪婪搜索方法.枚举搜素对所有特征组合进行检测,优点是结果准确,但计算量大,不适用于大规模数据;随机搜索随机地从所有位点中挑选位点子集,考察当前位点的重要性.重复以上步骤,尽可能覆盖所有位点.当位点总数大时,很难覆盖所有位点;贪婪搜索算法不挑选尽可能覆盖所有位点的多个位点子集,先通过某种标准过滤掉绝大部分噪音位点,再对剩余位点探索交互作用.

SNPHarvester算法[13]是一种基于启发式搜索的算法,利用路径选择过程来采样搜索空间.其基本思想是先通过单位点显著性检测删除有主效应的位点,在剩余位点中用爬山算法贪婪地搜索出一组互作效应最强的位点集合,最后利用惩罚回归从候选结果中提取交互作用.SNPHarvester计算速度快,每发现一个局部最优后,对应的位点集合就被从搜索空间中移除,搜索空间越来越小.其主要问题在于算法易陷入局部最优且不易并行化.
AntEpiSeeker算法[14]是一种基于蚁群优化的2阶段启发式搜索方法,可适用于大规模数据集中位点互作的检测.在筛选阶段,AntEpiSeeker用蚁群算法搜索含有多于给定位点数的SNPs子集,从中选出卡方统计量最大的若干子集作为候选子集,再选出信息素最大的若干位点作为候选位点.在候选子集与候选位点中,通过穷举搜索发现包含给定数目位点的集合,采用卡方统计量的p-值作为评价函数.以上2阶段搜索重复2轮.AntEpiSeeker对边际效应鲁棒性较好,但需要指定一系列参数(包括蚁群算法的参数和2轮迭代涉及的参数),算法性能受参数影响大.
2 基本概念与问题描述
2.1 基本概念
本节将给出文中所涉及到的一些基本概念并对要解决的问题给出形式化描述.

Fig. 1 An example of maximal allelic common subsequence图1 极大等位公共子序列的示例图

极大等位公共子序列与广为熟知的最长公共子序列不同.如图1所示,若S1=100101和S2=101011为任意2个由0和1构成的序列,则序列Sα=101为S1和S2的极大等位公共子序列,序列Sβ=10101为S1和S2的最长公共子序列.显然,Sα≠Sβ.计算多序列的最长公共子序列是典型的NP-难问题[1],时间复杂度相对于序列数量N是指数级别的,而由图1不难得知,极大等位公共子序列通过简单的位“与”计算即可获得,时间复杂度仅为O(N).因此,极大等位公共子序列的计算量远小于最长公共子序列.
由定义1可知,对给定的数据集D,其中所有任意k条序列的极大等位公共子序列的数量随着序列数|D|的增加而增加.一些极大等位公共子序列可能会存在较大的序列相似性.如果考虑所有的极大等位公共子序列,将引入很多重复计算,影响交互特征挖掘的执行效率.因此,提出基于序列多样性的特征区域选择方法.通过将问题规约到经典的最小独立支配集问题,设计有效的解决方法,使得在尽可能不丢失结果的前提下,减少很多重复计算.以下是最小独立支配集的具体定义.
定义2.最小独立支配集. 给定图G={V,E},其中,V和E分别表示该图的点集和边集.若存在V*⊆V,满足∀u,v∈V*,(u,x)∉E,并且∀w∈V,要么w∈V*,要么∃x∈V*,(x,w)∈E,则称V*为V的独立支配集(independent dominating set, IDS).进一步,包含节点数最少的独立支配集被称为最小独立支配集(minimum independent dominating set, MIDS).
本工作的主要任务是从大规模序列中挖掘与目标变量(如某种疾病或表型)存在显著关联的高阶交互特征,因此有必要先明确k阶交互特征的定义.
定义3.k阶交互特征. 令F为含有k个特征f1,f2,…,fk的特征子集,C为某个特征或特征子集与目标变量(类标签)相关性的度量.若满足对F的任一划分F={F1,F2,…,Fl},C(F)>C(Fi),其中i∈[1,l],l≥2且Fi≠∅,则称f1,f2,…,fk为k阶交互特征.
由定义3可知,如果f1,f2,…,fk为k阶交互特征,当且仅当其对目标变量(类标签)的影响大于其任一子集对目标变量(类标签)的影响.
2.2 问题定义
统计显著性是常用的特征质量度量标准.对给定的候选特征集Fi,基于单次假设检验即可判断其与类标签cr是否存在显著的统计关联.但是,大规模序列数据中的待检验k阶交互特征数量极其庞大,多重假设检验引起的假阳性结果增多问题会给后续的生物验证带来巨大的资源浪费.族错误率FWER(family wise error rate)是指挖掘结果中至少存在一个假阳性结果的概率P(FP≥1),其中FP代表总体结果中的假阳性个数.FWER≤α意味着总体挖掘结果中至少出现一个假阳性结果的概率被控制在α以下.FWER在总体假阳性结果控制方面有坚实的理论基础[15].因此,本研究要求挖掘结果满足给定的族错误率阈值α.
问题描述:给定包含m条序列S={s1,s2,…,sm}和t个类标签C={c1,c2,…,ct}的数据集D,记si={fi 1,fi 2,…,fi n}为序列si包含的n个特征,cj为si对应的类标签,其中1≤i≤m,1≤j≤t,发现:所有满足定义3,与特定类标签cr∈C存在显著统计关联的k阶交互特征并且满足FWER≤α.
3 PACOIFS框架
第2.1节所述,大规模生物序列中的高阶交互特征挖掘面临着密集计算问题.本节将提出一种新的基于并行处理和演化计算的解决框架(PACOIFS),并对构成框架的各主要步骤加以详细介绍.
3.1 整体思想
图2给出了PACOIFS框架的总体示意图:

Fig. 2 An illustration of PACOIFS图2 PACOIFS框架示意图
框架由4个主要步骤构成:
1) 数据预处理.对原始数据进行编码后,执行特征维度削减,提前过滤一些无关特征,提高整个框架的执行效率.
2) 数据划分.基于极大等位公共子序列MACS,将原始高维特征数据划分成一系列低维特征区域,并保证其“低耦合高内聚”性(即特征交互在区域内的可能性高,在区域间的可能性低).
3) 特征区域筛选.许多划分后的特征区域具有较高的相似度.如果在所有特征区域上都执行交互特征挖掘,会产生很多重复结果,降低计算效率.采用最小独立支配集的思想,基于多样性原则选择代表性的特征区域.
4) 交互特征挖掘.基于选定的代表模式对数据进行垂直分解,提出基于置换搜索的并行蚁群算法,进行高效且有效的交互特征选择.
3.2 基于块和位点过滤的数据预处理
原始SNP数据一般为2种形式:基因型或单体型数据.以某位点为例,若用A表示出现频率较高的等位基因,a表示出现频率较低的等位基因,则基因型数据有3种形式:AA,Aa或aa,单体型数据有2种形式:A或a. 为便于计算机处理,通常需要先对原始SNP数据进行编码,前者的3种状态分别编码为0,1,2,后者的2种状态分别编码为0和1. 提出的方法可同时兼容基因型数据和单体型数据.
理论上,对特定的复杂疾病,不可能所有的位点都与之具有遗传学关联[16]. 称基因组中与特定疾病具有真正遗传学关联的区域为“热区”.“热区”定位是影响交互特征发现效率和有效性的关键因素.很多方法通过采用基于单位点过滤的方法,保留可能的“热区”. 其问题在于,单位点过滤大多只保留具有强边际效应的候选位点,许多具有弱边际效应但具有强交互作用的位点组合被遗漏了.在对原始数据编码后,本节中将提出一种结合块过滤和位点过滤的2阶段过滤方法BLFilter. 先通过基于图论的块过滤,粗粒度地保留与疾病具有较高关联可能性的区域;再通过细粒度的位点过滤进一步提炼保留区域内的位点,以最大限度地保留边际效应弱但交互作用强的位点.

Fig. 3 The graph model of blocks图3 分块的图模型
3.2.1 块过滤
(1)
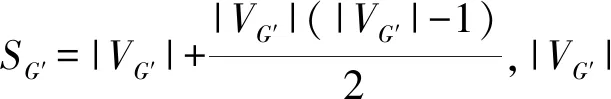
根据式(1),图G中的密集子图对应的区域分块内部和相互之间都存在较多的显著交互位点对.因此,直观上可理解为与疾病存在显著关联的“热点”区域,问题被转化为密集子图发现问题.目前,从图G中发现最密子图的最优算法时间复杂度为O(|V|(|V|+|E|)log(|V|)log(|V|+|E|))[17],虽然是多项式级的,但对大规模序列分析而言,仍是不现实的.现有的2-近似算法贪婪算法[18]没有考虑顶点和边带有权重的图.针对该问题,本节提出一种能有效处理边权重和点权重的密集子图发现算法GREEDYVED.该算法迭代地移除当前图中具有最小平均度数的顶点vx及相关的边,并计算移除后图G′的密度.整个迭代结束后,d(G′)最大的子图被作为最密子图输出.伪码如算法1所示,其时间复杂度为O(|V|+|E|).
算法1.块过滤算法GREEDYVED.
输入:权重图G=(V,E);
①Sn=V;
② fori=nto 1 do
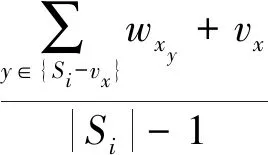
④Si-1=Si{vx};
⑤ end for

定理1对块过滤算法GREEDYVED的近似程度给出了证明.

(2)

证毕.
3.2.2 位点过滤
对保留区域内的位点,进一步执行细粒度的位点过滤.伪码如算法2所示:
算法2.位点过滤算法.
输入:块过滤后的位点集合X;
输出:位点子集X′⊆X.

② fori=2 to |X| do
then
max_pos=i并且保留max_pos之前的所有位点至X′;
⑤ end if
⑥ end for
⑦ fori=max_pos+1 to |X| do
then
⑨ 将Xi和Xj保留至X′;
⑩ end if
具体过程如下:

与传统方法相比,基于块和位点的2阶段过滤方法,既保持了过滤的高效性,又考虑了过滤的有效性.不仅保留了具有强边际效应的位点,而且最大限度的考虑了边际效应弱,但具有潜在交互作用的位点.
3.3 基于MACS的数据划分
大规模序列中的高阶交互特征挖掘是典型的计算密集型任务,位点数是制约交互作用挖掘效率的根本因素.现有并行框架大多对数据进行水平分组,从减少各节点处理的序列数量角度提高计算效率.与此不同,本节从减少各节点处理的位点数入手,提出一种基于MACS的数据垂直分解策略,保证交互作用划分后的“低耦合高内聚”性,即每个区域内的特征交互可能性高,不同区域间的特征交互可能性低.
3.3.1 划分粒度
如问题定义所述,本研究要求挖掘结果的族错误率FWER被控制在α以下.利用统计中的置换检验方法,理论上可以获得满足FWER≤α的最大单次假设检验阈值δ,但计算量极其巨大.Llinares-López等人于2015年提出了一种基于假设检验的高效显著模式挖掘算法FastWY[15],一种快速确定δ的计算方法,并证明了模式的支持度γ与显著性p值之间的对应关系.

Table 1 p_value based on the Contingency表1 基于列联表的p值计算

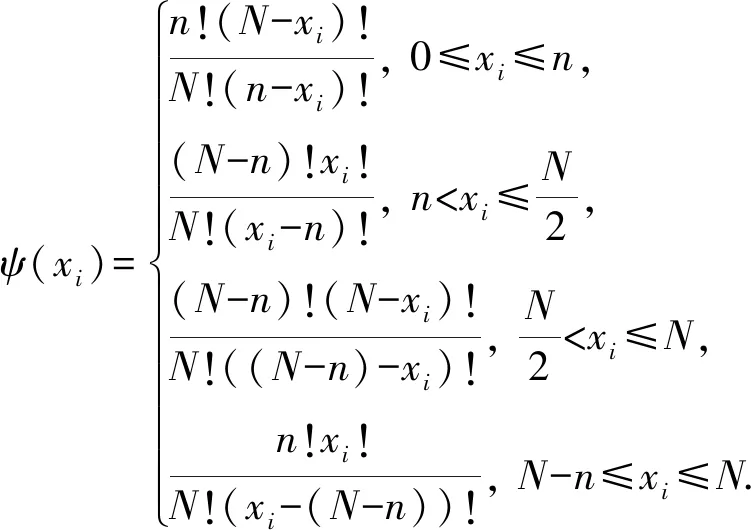
(3)
进一步,证明了最小可达p值的下界ψ*(xi)相对于xi是单调递减的.利用FastWY快速确定δ之后,若记其对应的xi为ψ*-1(δ),则只需检测所有出现次数不低于ψ*-1(δ)的特征集合.根据定义1,数据集D中任意ψ*-1(δ)个序列的MACS构成的子序列集合M即为划分后的交互特征候选区间.注意:M中的任一序列s在D中的支持度至少为ψ*-1(δ),支持度不低于ψ*-1(δ)的模式或者为M中的序列或者为M中序列的子序列.因此,显著交互特征只可能存在于M中的每个序列内部,不同序列之间的位点不可能存在显著交互作用.
3.3.2 基于MACS的并行划分
目前,任务规约为求数据集D中任意ψ*-1(δ)个序列的MACS构成的子序列集合M的问题.本研究中利用基于MapReduce的并行框架来高效地求得结果.
3.4 基于MIDS的特征区域筛选
基于交集操作得到的MACS数量随着数据行数的增多而显著增多,在大规模序列数据分析时对整个算法的挖掘效率影响非常大.在实验中发现了一个现象,即许多MACS具有较高相似性.如果不加处理,直接基于所有的MACS执行挖掘过程,会产生大量冗余的计算.因此,在尽可能保证结果完整性的前提下对得到的特征区域进行多样性选择.目的是选择尽可能少的代表性区域,使其相互间差异大,但与未选择的非代表性区域有较大的相似性.
该任务可以规约为最小独立支配集(MIDS)问题.每个MACS作为图G中的1个顶点,如果MACS间的相似度大于给定阈值,2个顶点间有边存在.提出了基于贪心策略的(greedy,relevance,diversity,coverage, GRDC)算法进行特征区域多样性选择.基本思想如下:每次从无向图中选择度最大的节点并将其加入最终结果集中,然后将与该节点存在连接的边删除掉,迭代至无向图G中边为空为止.
进一步,实现了基于MapReduce的贪心策略GRDC.在Map处理阶段,为了提高GRDC算法的运行效率,先将每行数据的对应MACS集合进行特征区域多样性选择,得到若干代表此行序列的MACS子集.因为当特征维数比较高时,每行数据对应求交得到的MACS数量相对也比较大,可以先在每行数据对应的集合内部进行多样性选择,使得最后进行多样性选择的MACS数量大大减少.然后,依次对原始数据集中每一行数据对应的MACS集合都执行此操作.在Reduce处理阶段,先得到对应Map任务处理后的结果,然后将汇总得到的MACS集合再进行一次特征区域多样性选择,得到最终经过多样性选择后的特征区域MACS集合,并将其写入到HDFS中.
3.5 基于置换搜索的并行蚁群交互特征挖掘
本节将在分解后的特征区域内,利用蚁群算法发现k阶交互作用.
3.5.1 基本蚁群算法
本节引入面向k阶交互的基本蚁群算法.其具体步骤如下:
首先,初始化antNum个蚂蚁对象和迭代次数iterNum.然后,每个蚂蚁随机选择一个初始特征,原始数据集中的每个特征对应一个初始信息素值τ0.每一个蚂蚁根据式(4)选择下一个特征,直到选择出的特征子集大小为k.比较每只蚂蚁搜索得到的显著特征子集,选择出显著性特征子集的并根据式(5)更新集合中每个特征的信息素浓度大小.
(4)
τk(t+1)=(1-ρ)τk(t)+Δτk(t),
(5)

3.5.2 置换策略
基本蚁群算法在每一次蚁群迭代过程中,得到的显著交互特征子集信息不能够保存下来.在随后的迭代过程中,是根据每个特征位点的概率密度进行重新选择的.针对此问题,提出基于置换的蚁群算法,使得改进后的蚁群优化算法能够避免显著特征所带来的边际作用影响.基本思想为:保留上一步迭代过程中得到的显著交互特征子集,然后每次对集合内的一个特征进行置换.在选择下一个特征时,根据每个特征已置换次数选择集合外的一个特征与集合内的每个特征进行置换,从而减少特征的重复选择增加多样性.如果新的特征子集较原来的显著,就执行此次置换,否则不执行.这种方法,能够很好地利用高阶交互特征子集信息,有效防止低阶特征所带来的边际作用影响,从而得到显著的高阶交互特征子集.本文中,该策略被记为RouteSearching,共包括3个阶段:
1) 根据式(4)选择大小为k的初始位点数.在初始位点选择过程中,根据计算每个位点对应打分值Score,然后计算出对应的显著性p_value,最终选择出大小为k的位点子集.
2) 对迭代次数以及是否执行下次置换标记进行初始化,并通过置换搜索的思想,迭代地选出显著位点子集,并将其全部加入到集合S′中.当已选位点子集外没有使该特征子集更显著的位点时,迭代停止.
3) 对集合S′中的每个显著位点子集进行特征位点的后向检测,目的是为了删除掉冗余特征子集,最终得到显著的不大于k阶的交互特征子集集合S.
由于空间有限,此处略去RouteSearching的具体实现伪代码.
3.5.3 基于置换的蚁群算法
在基本蚁群算法的迭代搜索中,加入置换策略RouteSearching后,得到改进的蚁群算法,步骤如下:
步骤1. 计算数据集中每个位点的Score,选择出显著位点,将其移除. 这样能够减少强相关特征所带来的边际作用的影响.
步骤3. 通过置换策略RouteSearching选出显著交互特征子集集合S′,并将其加入到结果集合S中.
步骤4. 对步骤3得到的集合S′中的每个显著特征位点进行信息素大小更新操作.然后进入到下一个蚂蚁迭代搜索中,直到所以蚂蚁迭代搜索完成,算法终止.
3.5.4 并行蚁群算法
在本节中,基于MapReduce模型对基于置换的蚁群算法进行了并行化,提出了相应的算法PACOIFS.以下将给出详细的描述.
蚁群优化算法从本质上来说就属于一种并行算法,具有比较高的可并行性.在蚁群中的每只蚂蚁搜索的过程其实就是得到可行解的过程,蚂蚁之间搜索过程是相互独立的,每只蚂蚁搜索可行解的过程只与信息素浓度大小以及启发式函数有关,并且当整个种群迭代搜索完成之后,才进行一次全局信息素浓度大小的更新操作.因此,可以把每个蚂蚁放到不同的机器节点上并行地搜索可行解,这样就使得集群并行化运行的优势得到了充分利用.蚁群算法比较常用的并行方式有2种:1)并行交互蚁群,即是将整个蚁群划分为几小部分,然后在这些部分蚁群之间并行;2)并行蚂蚁,即是将整个蚁群中的每一个蚂蚁进行并行.通过以上介绍可以发现,并行蚂蚁方式其实是并行交互蚁群方式的一种极端化形式,即是将整个蚁群划分成蚂蚁数目个小部分蚁群.由于并行交互蚁群方式每台机器所对应Mapper任务迭代时间会很长,在数据集规模比较大的情况下对机器性能要求较高.本文将采用并行蚂蚁方式,因为这样能够将计算任务更好地分配到各个机器节点上,减轻每台机器的计算压力.伪码如算法3所示.
算法3.PACOIFS().
Map Function(key,value=第k个蚂蚁对应的交互特征子集集合Sk).
输入:SNP数据集、信息素以及蚁群参数等信息;
输出:〈key,value=Sk〉.
① 读取数据集D并划分成m块;
② for 每个块bido
③ 读取信息素及蚁群参数等信息进行初始化;
④Sk←RouteSearching(bi,α,k,maxQ,SNPs);
⑤ 输出〈key,value=Sk〉.
⑥ end for
Reduce Function(key,value=显著交互特征子集集合S).
输入:〈key,Sk〉;
输出:〈key,S〉.
① for 在Map任务中的每个Skdo
② 添加SktoS;
③ end for
④ for 每个S中的SNP do
⑤ 根据式(5)更新每一个SNP位点的信息素;
⑥ end for
⑦ 将更新后的信息素向量信息写入到HDFS之上;
⑧ 输出〈key,S〉.
根据本文中所采用的蚁群并行方式可知,在MapReduce框架中,局部搜索的过程即对应着Map接口,全局信息素浓度大小更新操作即对应着Reduce接口.每个MapReduce Job即对应一次蚁群进行迭代搜索交互特征子集的过程,其中每个Mapper任务即对应着一组蚂蚁搜索交互特征子集的过程,有多少组蚂蚁就会对应多少个Mapper任务,每一个Reduce任务即是将Mapper任务结果汇总并写入到HDFS中以及信息素更新操作的过程,整个蚁群的迭代即是MapReduce Job的迭代.由于在MapReduce框架下开启Mapper或者Reduce会有相应的自身开销,本文将会使用少量的蚂蚁数目,把整个算法的迭代搜索尽量放到每个Mapper任务中去,3.5.2节给出的置换策略恰恰符合这种需求,因为本文是基于每个蚂蚁在迭代搜索过程中通过置换的思想来挖掘交互特征子集的.
4 实验与分析
4.1 实验环境
本实验所用的软硬件环境是集群由5台服务器组成,每台服务器的配置为Red hat 64位操作系统,16核CPU,主频1.9 GHz,16 GB内存,2 TB硬盘;Hadoop版本为2.6.0,Spark版本为1.6.0,Java版本为1.8.0,Scala版本为2.10.4.单机开发环境:操作系统为Windows7 32位旗舰版,主频3.10 GHz,4 GB内存,500 GB硬盘;开发工具IntelliJ IDEA Community Edition 15.0.2,Java版本为1.8.0,Scala版本为2.10.4.
4.2 数据集
1) 模拟数据集.采用由Marchini等人[19]提出的相加(模型1)、倍增(模型2)以及阈值(模型3)三种基因模型.通过设置各个模型中不同的参数值来产生相应的实验测试数据.模拟数据集中共有4 000个样本(其中包含2 000个病例和2 000个对照样本)和2 000个SNP位点.由于数据的统计能力受MAF的影响较大,取其值为0.1,0.2,0.5;边际作用λ取值较小,在模型1中设置大小为0.3,在模型2以及模型3中都设置为0.2;患病率(Prevalence)在每个模型中取值分别为0.1,0.4,0.7;对于连锁不平衡r2大小,每个模型分别取值0.7和1.此外,还通过增加每个模型对应数据集中的数据行数和SNP位点数来检测算法在大数据集中的统计能力.
2) 真实数据集.使用了糖尿病患者数据作为真实数据集.该糖尿病患者数据集从美国凯斯西储大学获取.该数据集共包含有90个样本个体,其中包括45个日本人和45个中国人.日本人中有38个患病(case)样本,中国人中有11个患病(case)样本,其他都为未患病(control)样本,并且每个样本都由22条染色体组成,每条染色体所对应的SNP位点个数在3 000~20 000之间,详细信息如表2所示:
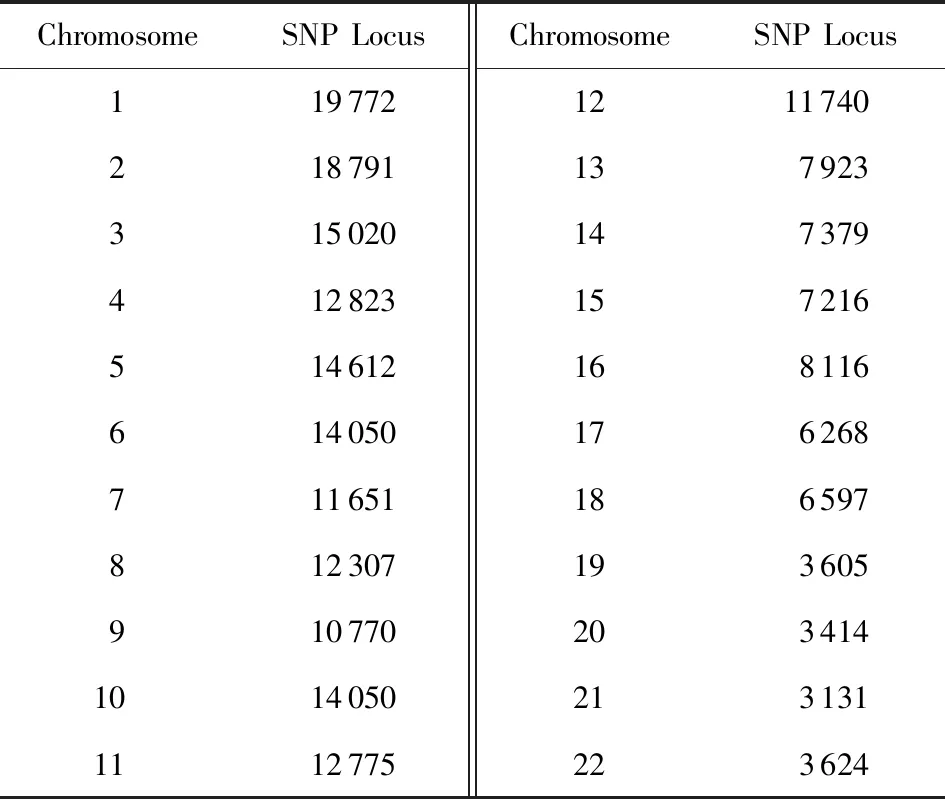
Table 2 The Diabetes Dataset 表2 糖尿病数据集

Fig. 4 Power comparison on MAF change for different models图4 不同模型的识别力Power受MAF变化比较图
4.3 参数设置
本文算法中主要包含参数:统计显著性阈值α、待选择SNP子集大小k、局部搜索算法Route-Searching迭代次数上限值maxQ、蚁群种群大小antNum、种群迭代次数iterNum、SNP位点初始信息素大小τ0以及信息素挥发系数ρ.
统计显著性阈值α通过多重假设检验校正设置为α=0.01,每个SNP位点的初始信息素大小τ0=1,信息素挥发系数ρ=0.05,待选择SNP位点集合大小k∈[1,5].由于在置换搜索RouteSearching中循环条件由当前迭代次数和是否执行下次置换参数共同决定,为了防止每个蚂蚁在局部置换搜索中迭代次数过多,并且又能够利用已经得到的显著SNP位点集合信息,将最大迭代次数设置为maxQ=[0.01,0.2],默认值100.蚁群种群大小antNum∈[0.01,0.5]×N,种群迭代次数iterNum∈[0.05,0.7]×N,蚁群算法中2个重要的参数是种群大小antNum和种群迭代次数iterNum.
4.4 识别能力分析
本节将在模拟数据集上对提出的算法进行识别能力分析.算法识别能力是指算法能够正确检测出致病位点的数据集个数在所有数据集中所占的比例大小,用式Power=NαN表示.其中,N表示每个模型中对应的数据集的数目,Nα表示算法能够正确识别出致病位点的数据集数目.根据4.3节中介绍的各基因模型参数设置,生成对应的模拟数据集,每种模型对应18种不同参数组合.实验中将要对比的参数分别为次等位基因频率MAF、患病率Prevalence以及连锁不平衡r2.基于各模型对应的数据集对文中算法PACOIFS进行实验测试,并将实验结果和代表性算法SNPHarvester[13]、AntEpiSeeker[14]以及贝叶斯上位关联映射(BEAM)[12]进行对比分析.具体实验结果分别见图4~6所示.
图4中,参数MAF的变化和模型的不同对各个算法的识别能力的影响还是较大的.其中,对于本文算法PACOIFS和AntEpiSeeker算法大致是随着MAF的增大其Power随之增大,表明当增大致病SNP位点的次等位基因频率,MAF会增大算法对致病SNP位点集合的检测能力.这2个算法在3个模型中的识别能力大致相同,都可以达到不错的效果.算法BEAM,SNPHarvester的波动在图4(a)和图4(c)下不太稳定,在图4(b)下是恰好呈相反的趋势,即是随着MAF的增大其Power反而减小,即是表明当增大致病SNP位点的次等位基因频率MAF会减小算法对致病SNP位点集合的检测能力.
图5中,患病率Prevalence的变化对算法Ant-EpiSeeker和BEAM的影响不太大,大致在图5(b)和图5(c)上呈轻微上升趋势,而在图5(a)中当Prevalence=0.4时性能较差,总体上算法Ant-EpiSeeker要比算法BEAM的Power性能好一些.本文算法PACOIFS在各模型中则表现出和算法AntEpiSeeker相似的趋势,其Power大致上比算法AntEpiSeeker好一些.算法SNPHarvester则表现出和其他算法不一样的性能,其在图5(a)和图5(b)中当Prevalence=0.4时性能较差,在图5(c)中Power随着Prevalence增大而呈上升趋势.

Fig. 5 Power comparison on Prevalence change for different models图5 不同模型的Power受患病率变化比较图
图6中,算法BEAM,SNPHarvester,AntEpi-Seeker以及本文算法PACOIFS在图6(a)~(c)中的Power值都随着连锁不平衡r2的增大而提升.其中,算法AntEpiSeeker和本文算法PACOIFS表现较其他算法好,在图6(b)中这2个算法都达到了最好值Power=1.在图6(c)中的Power性能比图6(a)和图6(b)稍差,并且算法BEAM,SNPHarvester大致也呈这种趋势.
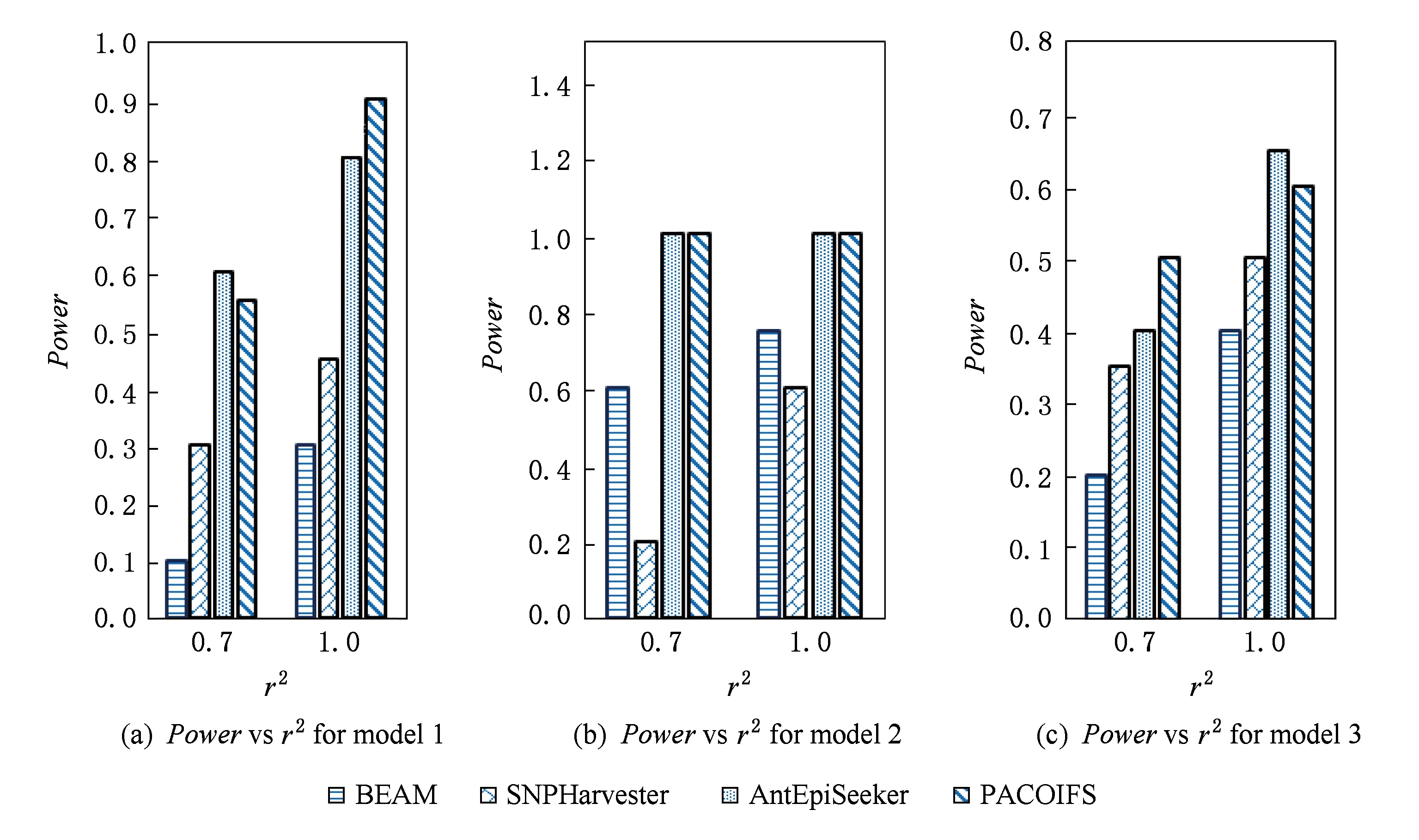
Fig. 6 Power comparison on r2 change for different models图6 不同模型的Power受r2变化比较图
通过设置不同的参数,即蚁群种群大小和迭代次数,对本文算法PACOIFS进行相应的识别能力Power对比分析.首先,生成图6(b)默认参数下规模为2 000×2 000的模拟数据集,然后设置种群大小antNum=100,iterNum分别为200,250,300,350,400,迭代次数iterNum=200,antNum分别设置为100,150,200,250,300,将实验结果和算法AntEpiSeeker在相同实验参数下进行对比分析.结果如图7~8所示:
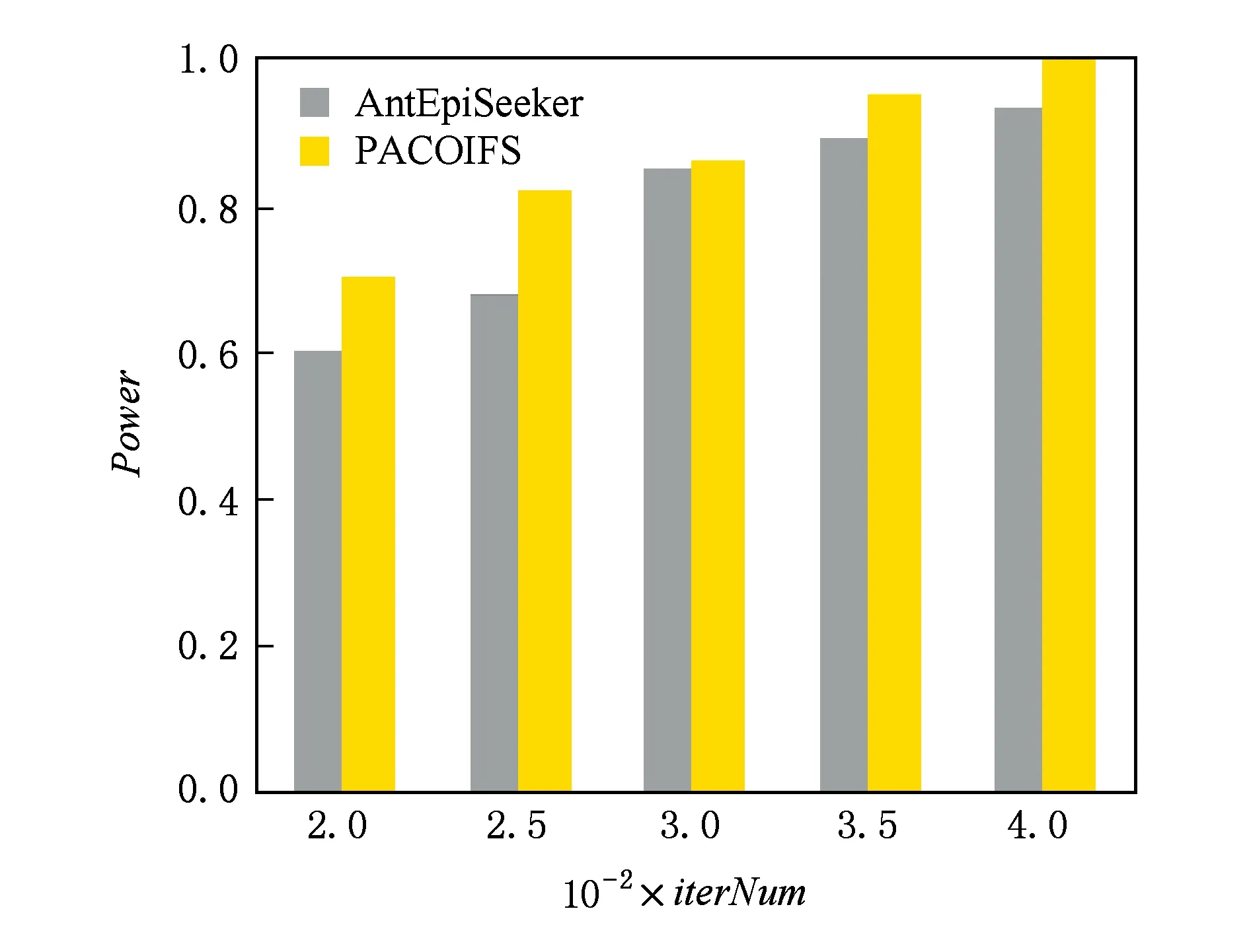
Fig. 7 Power comparison on iterNum change图7 Power受iterNum变化比较图
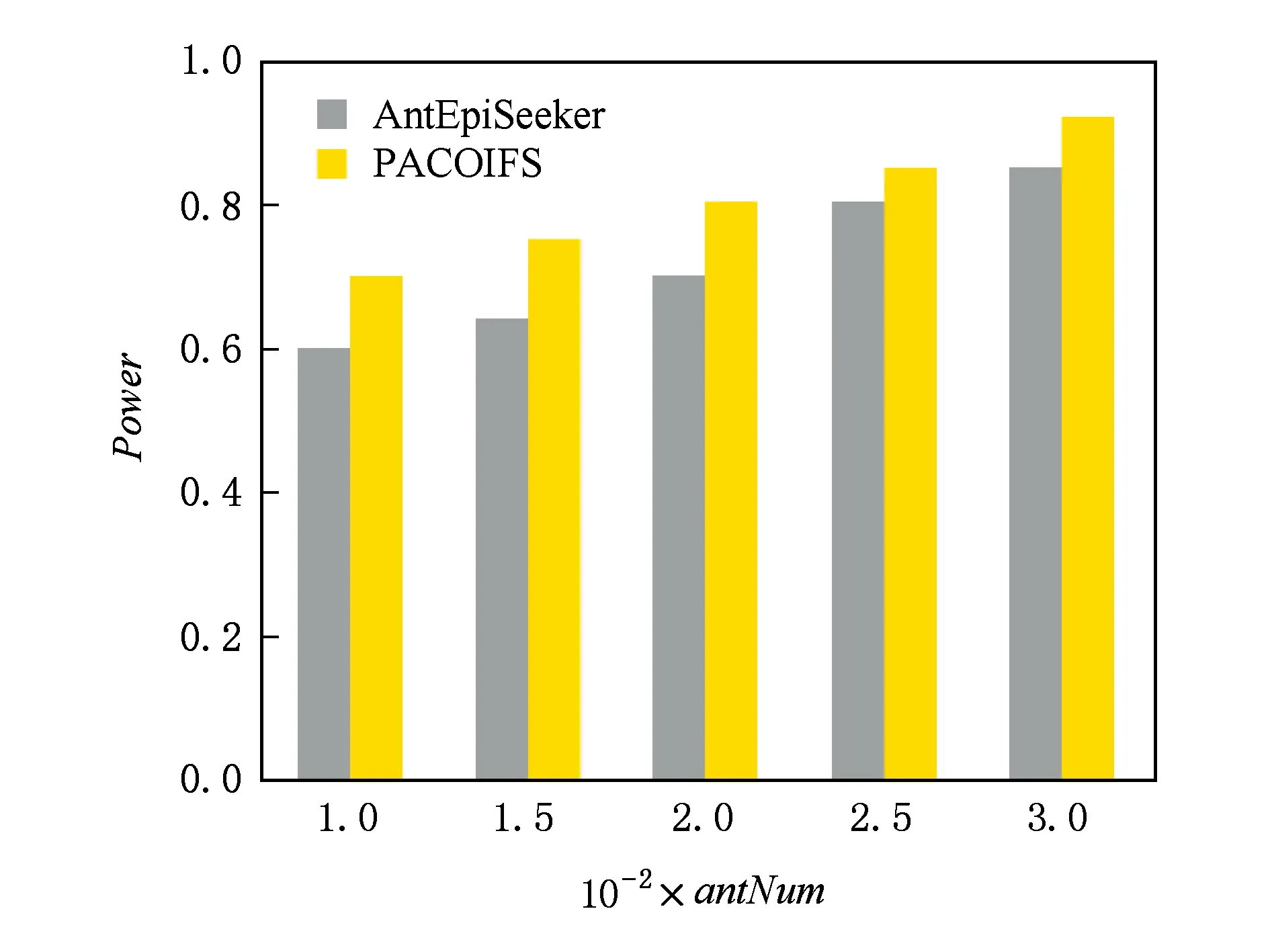
Fig. 8 Power comparison on antNum change图8 Power受antNum变化比较图
4.5 可扩展性分析
本节将利用模拟数据集对算法PACOIFS进行可扩展性方面的分析.首先,观察分析提出算法随数据行数及SNP位点数的增加,其Power性能的变化,并将实验结果与SNPHarvester[13],AntEpiSeeker[14],BEAM[12]进行对比.基于基因模型2的实验结果分别如图9和图10所示.
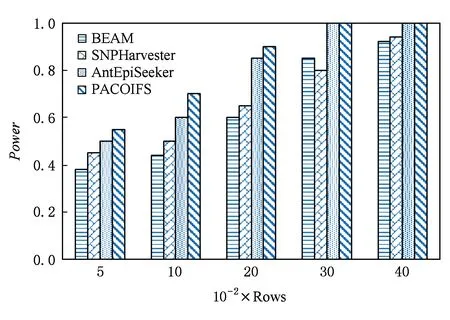
Fig. 9 Power comparison on datalines change图9 Power受数据行数变化比较图

Fig. 10 Power comparison on SNP-locus change图10 Power受SNP位点数变化比较图
图9中,算法BEAM,SNPHarvester,AntEpi-Seeker及算法PACOIFS在模型2中的Power性能随数据行数的增加而增大.其中,算法AntEpi-Seeker和本文算法PACOIFS在SNP位点数为2 000、数据行数为3 000和4 000时均达到了最好性能.因为随着样本数的增多其统计能力也会随着增大,并且随着数据行数的增加本文算法要比算法AntEpiSeeker的总体Power性能要好.图10中,算法BEAM,SNPHarvester,AntEpiSeeker以及算法PACOIFS在模型2中的Power性能随着SNP位点数增加而大致减小,其中算法AntEpiSeeker和算法PACOIFS在数据行数固定为2 000对应SNP位点数为100的时候均达到了最好性能.当SNP位点数逐渐增加的时候本文算法PACOIFS也是呈最好的Power性能,因为本文在算法框架加入了特征区域划分阶段,使得SNP位点的增加对其算法性能影响不大.这也是大多数算法在高维数据中性能表现不好的原因.
算法随着SNP位点数增加的响应时间变化情况如图11所示.由于真实情况下,数据集中的样本量一般很少而SNP位点数一般会很大,所以这里只分析随着SNP位点数的增加,各算法在时间效率上的变化情况.同样是在基因模型2上采用不同位点数进行实际测试,其中样本数为2 000.由图11可见,算法BEAM和SNPHarvester相对比较快,而算法AntEpiSeeker和本文算法较慢.这是因为算法AntEpiSeeker加入了后续最小化假阳性处理阶段,并且其使用了2阶段蚁群迭代,所以时间相对较长.但是,本文算法ACOIFS由于加入了无关特征削减、序列之间求MACS等前期处理过程,所以运行时间也比较长,这也是其Power性能比较好的原因.随着SNP位点数增加,算法BEAM和SNPHarvester的响应时间增长幅度较明显,而算法AntEpiSeeker和本文算法的响应时间虽然也在增加,但并不明显.并行算法PACOIFS表现最好,在SNP位点数为1 000的时候其运行时间已经达到最少,并且随着SNP位点数增加,这种趋势更加明显.

Fig. 11 Runtime comparison on SNP-locus change图11 运行时间受SNP位点数变化比较图
4.6 加速比分析
加速比是同一个任务在不同处理器或并行处理器中运行时间的比值,用来衡量程序代码并行化或并行系统的真实有效性.在独占CPU资源的前提下,假设某个串行程序运行时间为Ts,而将该程序并行化后,k个进程在k个CPU并行运行的时间为Tk,那么程序并行化之后的加速比R则为R=TsTk.由于在本文算法框架中包括特征区域划分以及特征区域选择阶段,为的是将原始长的序列切分成若干短的序列并对其进行多样性选择,这样同时也使得数据易于并行化处理.将在真实数据集上采用不同SNP位点数来测试算法PACOIFS基于MapReduce框架的加速比,结果见图12所示:
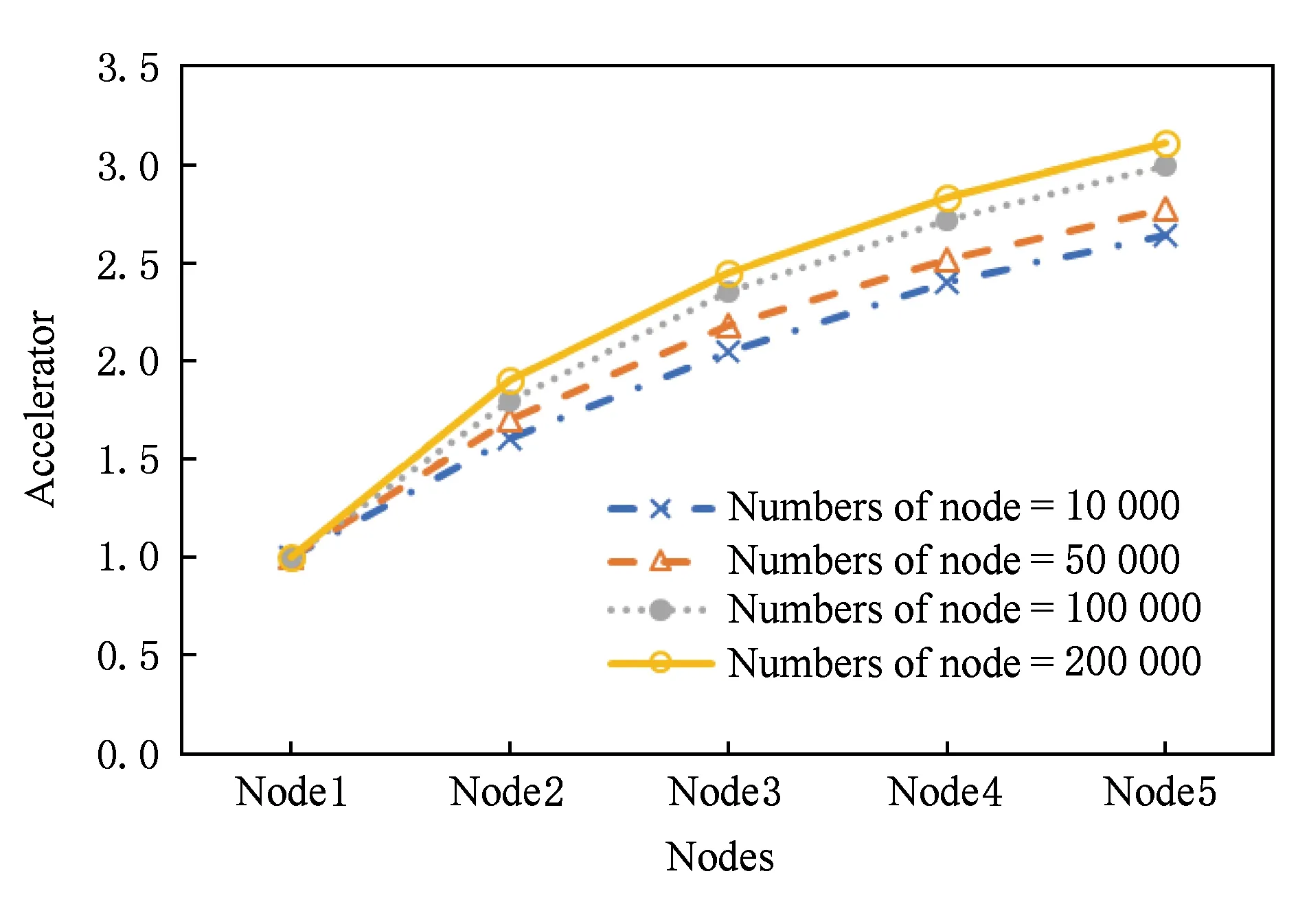
Fig. 12 Speed up on different datasets and nodes图12 不同数据量不同节点个数的加速比
由图12可见,本文算法的加速比随着节点个数的增加逐渐增加,同时增加的趋势在不断减小.也就是说,如果不断地增加节点,则处理时间不会呈线性的减少.这主要是因为MapReduce框架下,数据传输会消耗时间.由MapReduce模型本身的特点可知,Map任务完成后,需要将处理结果进行排序,并根据partition函数将数据传输到对应的Reduce节点上,即shuffle处理阶段,这段时间需要进行数据拷贝.当数据集较大时,增加节点虽然能使数据处理的时间减少,但并没使得数据传输的时间减少.因此,加速比曲线没有呈斜率为1的趋势增加.
4.7 有效性分析
根据以上实验结果,算法AntEpiSeeker和本文算法PACOIFS性能表现最接近,因为AntEpiSeeker使用了2阶段蚁群迭代搜索并加入了后续最小化假阳性处理阶段,而本文算法由于加入了无关特征削减以及特征区域划分求MACS等前期处理过程,所以实验结果相对较好.在算法有效性分析中,将基于糖尿病患者数据集对PACOIFS和AntEpiSeeker进行比较测试和对比分析.首先,根据算法AntEpi-Seeker中的建议将实验参数设置为iItCountLarge=10 000,iItCountSmall=20 000,其他参数取默认值;本文算法参数设置为antNum=2 000,iterNum=20 000.
表3中列举出了2种算法识别出的部分显著位点集合.可以看出,本文算法PACOIFS所识别出的显著位点集合比算法AntEpiSeeker数目多,而且结果更显著.由于本文算法在开始时将比较显著的单SNP位点提前移除掉,因此得到的结果更好地反映出了在全基因组范围内SNP位点交互作用分布的情况.最后,将选择出的结果和糖尿病的参照致病位点集合进行对比,进一步验证了结果的可靠性.

Table 3 Some Significant SNP Groups Identified by AntEpiSeeker and PACOIFS表3 AntEpiSeeker和PACOIFS识别出的部分显著SNP位点集合
5 结 论
针对大规模生物序列中高阶交互特征挖掘面临的密集计算问题,本文提出了一种新的基于并行处理和演化计算的解决框架.与现有框架不同,提出的框架从减少分组数据位点数入手,对数据进行垂直分解.由于位点数是制约互作挖掘效率的根本因素,提出的框架具有天然的效率优势.同时,提出了极大等位公共子序列MACS的概念,能保证划分后交互作用的“高内聚低耦合”性,避免了分解后计算任务的耦合,减少了结果的丢失.在模拟数据集和真实的糖尿病患者数据集上,从识别能力、可扩展性、加速比、有效性等角度,将PACOIFS与相关经典算法进行了比较分析,结果表明提出的算法具有明显的效率和有效性优势.由于本文提出的算法框架采用求MACS的方式对特征区域进行划分操作,当样本较多时比较耗时,今后可以考虑利用已得到的结果减少求MACS的操作.另一方面,本文是在得到所有求MACS后进行多样性特征区域的选择,未来可以考虑选择一种在线处理方式进行多样性特征区域选择,以提高效率.
