核磁共振内标法测定磷酸伯氨喹片中磷酸伯氨喹的含量∗
2019-05-13王斌张永红刘文涛刘晨江
王斌,张永红,刘文涛,刘晨江†
(1.新疆大学理化测试中心,新疆乌鲁木齐830046;2.新疆大学化学化工学院,新疆乌鲁木齐830046)
核磁共振定量分析(qNMR)方法是以结构分析为基础的分析方法,具有简便快速、对样品无破坏性、重现性好、无需待测物的高纯标准品等优势[1−4].随着现代超导高磁场的脉冲Fourier变换核磁共振谱仪的应用,qNMR方法的灵敏度、准确度、精确度及分析速度等方面已达到或接近高效液相色谱的水平[5],是解决无标样定量分析问题的重要途径.近年来,qNMR 方法在化学化工、医药生物、农业食品和代谢组学等领域已得到广泛应用[6−13],中国药典于2010 版开始将该技术作为法定标准收载于通则( 附录)中[14,15].因此,qNMR方法应用于药物定量分析已成为趋势,是鉴别假冒、伪劣药品的有效方法,对管理我国的药品市场也有着重大的意义.
疟疾是目前全球倍受关注的三大疾病之一.非洲、中南美洲和东南亚的多数国家和地区是疟疾的高流行地区,尤其是恶性疟流行仍较严重.磷酸伯氨喹片是一种抗虐药,用于防治间日疟、三日疟的复发和传播[16−19].目前,在非洲及东南亚等疟疾多发地该药仍被广泛使用,在国内也被作为战略物资,其原料及片剂一直被保留生产.磷酸伯氨喹片有关物质的测定,各国药典方法也各不相同,《中国药典》2010年版采用的是薄层色谱法(TLC),该方法精确度差,且主斑点与主要杂质喹西特斑点分离度不够;《英国药典》2015年版采用正相高效液相色谱法(NP-HPLC),该方法操作繁琐,使用溶剂为挥发性有机溶剂,因此配制的溶液容易挥发造成误差,分离效果及检测灵敏度也差;《美国药典》38版采用反向高效液相色谱法(RP-HPLC),该方法操作较为简便,分离效果尚可,准确度高,但需要将药品进行预分离处理,除去药品外层包裹的糖衣,且需要磷酸伯氨喹标准对照品.本文在对比各国药典标准方法的基础上,建立了一种核磁共振内标法,简单、快速并且无需对照品测定磷酸伯氨喹片中磷酸伯氨喹含量的方法.
1 实验部分
1.1 仪器与试剂
仪器:Varian Inova-400 MHz超导核磁共振波谱仪(美国VARIAN安公司,DMSO-d6为溶剂,TMS为内标);KQ-100型超声波清洗器(中国昆山市超声仪器有限公司);ER-182A型电子分析天平(日本AND公司).
试剂:磷酸伯氨喹片(某制药公司,规格:13.2 mg,批号:110601);磷酸伯氨喹标准品(试剂级,纯度98%,上海阿达玛斯试剂有限公司);氘代二甲基亚砜(美国CIL公司,氘代度99.8%,0.03% V/V TMS);对苯二酚(试剂级,纯度99%,上海阿达玛斯试剂有限公司).
1.2 实验方法
1.2.1 试样准备
准确称量一颗磷酸伯氨喹片(100.8 mg),用玛瑙研钵研磨成粉末,混合均匀,称取上述过程处理的药品粉末20.4 mg加入核磁样品管中,然后加入1.1 mg内标物对苯二酚,再加入DMSO-d6溶剂振荡溶解.
1.2.21H-NMR测试条件
测定温度为50℃,检测频率为399.74 MHz,谱宽为6 999.70 Hz,90o脉冲宽度为10.80µs,采集时间为3.74 s,延迟时间为10.00 s,采集次数为100次.图谱处理时手动进行相位校正和基线校正,对内标物和样品的定量峰分别进行5次积分,取其平均值,得到积分结果.
2 结果与讨论
2.1 氘代溶剂的选择
称取多份20.0 mg经过研磨的磷酸伯氨喹片粉末,分别加入氯仿、甲醇、丙酮、二甲基亚砜、水等常用溶剂0.5 mL,通过振荡、超声辅助溶解10 min后,发现二甲基亚砜对样品的溶解性最佳,并且通过试验内标物对苯二酚也易溶于二甲基亚砜,因此选择氘代二甲基亚砜为溶剂.
2.2 内标物质的选择
内标物应具有含量准确、纯度高、性质稳定、溶解性良好、不与待测样品相互作用或反应,其定量峰与样品的信号峰不重叠等特点.内标物与待测物制成混合溶液,通过核磁谱图上相应定量峰的积分信号强度的比值可确定待测物的含量[1].常用的内标物有邻苯二甲酸氢钾、顺丁烯二酸、反丁烯二酸、对苯二甲醛、对苯二酚、苯、苯甲酸、3,5-二硝基苯甲酸、2,3,5-三碘苯甲酸、2,4-二硝基甲苯、3,4-二硝基甲苯、二氧六环等[20].根据内标物质在选定溶剂DMSO-d6中的溶解性和MestReNova软件模拟内标物和待测样品的核磁氢谱,内标物对苯二酚的两组峰均为单峰,且与待测样品的谱峰没有重叠,与定量峰的间距也较为合理,故选择对苯二酚为内标物.
2.3 1H-NMR谱峰归属
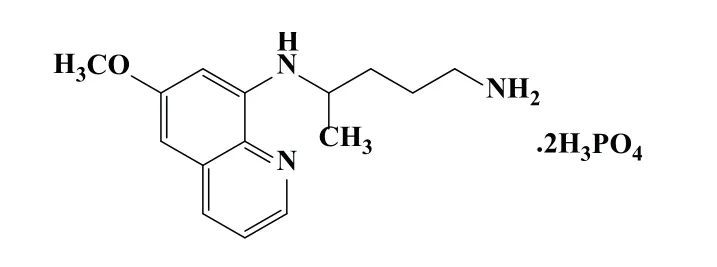
图1 磷酸伯氨喹分子结构Fig 1 Molecular structure of primaquine phosphate
分别对磷酸伯氨喹标准品、内标物对苯二酚、磷酸伯氨喹片粉末和内标物混合样品进行1H-NMR检测,对比图2中各图.磷酸伯氨喹的1H-NMR谱峰归属为δ:1.22 (s,2H,NH2),1.24 (d,3H,CH3),1.65(m,4H,2CH2),2.79 (t,2H,CH2),3.65 (m,1H,CH),3.83 (s,3H,OCH3),6.13 (s,1H,NH),6.29 (d,1H,CH),6.48 (d,1H,CH),7.42 (dd,1H,CH),8.07 (dd,1H,CH),8.53 (d,1H,CH),磷酸分子中的6个氢和DMSO-d6溶剂中的残留水的质子之间产生氢键作用,在δ:4.2∼5.7处产生了一个宽峰.内标物的1HNMR谱峰归属为δ:6.55(s,4H,CH),8.57(s,2H,2OH).图2中C谱图在δ:3.0∼4.0处出现的几组峰为药片糖衣中的糖成分所出的谱峰,δ:4.4∼5.8处出现的宽峰为磷酸分子中的6个氢、内标物中的2个OH活波氢和DMSO-d6溶剂中的残留水的质子之间产生氢键作用所产生的谱峰,由于三种不同氢核的氢键作用,化学位移较标准品和内标物发生了一定变化.
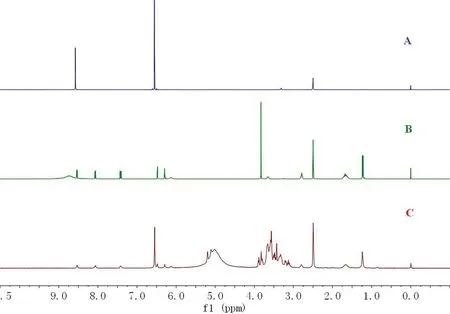
图2 1H-NMR谱图Fig 2 1H-NMR spectrum
2.4 定量峰的选择
定量峰应满足在选定的积分范围内无干扰,在此基础上应选择比较规则的信号峰以减小积分误差.如图2所示,内标物对苯二酚δ:6.55处为尖锐单峰,在磷酸伯氨喹药品分子中,δ:7.42、8.07、8.53处分别有一个峰型规则的峰,并且和内标物的峰之间没有重合,相互没有干扰,因此理论上均可以作为定量峰计算含量.但实际测试结果(见表1)表明:以化学位移在δ:8.07处作为定量峰测试的结果相对误差为0.84%,误差最小,因此选择化学位移在δ:8.07处的峰作为最佳定量峰.
2.5 回收率试验
采用加标回收法,在已知含量的样品中加入适量的磷酸伯氨喹标准品每个添加水平测定3次按1.221H-NMR测试条件测定计算回收率,结果见表1.由表1可以看出,磷酸伯氨喹的回收率在97.8∼102.7%之间,平均回收率为99.8%.表明该方法重复性良好,该结果证实所建立的qNMR方法能够应用磷酸伯氨喹片中磷酸伯氨喹含量测定.

表1 qNMR 方法回收率试验结果(n = 3)Tab 1 Recovery test results of qNMR method
2.6 磷酸伯氨喹定量分析结果
根据样品和内标物分子配制时质量、定量峰的质子数及核磁谱图中定量峰的积分面积比值,可以测定样品中磷酸伯氨喹的百分含量Ws,计算公式如下:

其中As是被测样品定量峰的积分面积,ns是被测样品定量峰包含的质子数量,Ms是被测样品的分子质量,Ar是内标物定量峰的积分面积,nr是内标物定量峰包含的质子数,Mr是内标物的分子质量,mr是加入的内标物质量,Wr是内标物的纯度,ms是样品质量.
测试药品重100.8 mg,标示磷酸伯氨喹含量为13.2 mg,因此磷酸伯氨喹含量为13.10%.实际测试计算结果见表2.结果表明:分别以δ:7.42、8.07、8.53的三个单质子峰作为定量峰计算得到的磷酸伯氨喹含量Ws为12.86%、12.99%、13.25%,该方法的相对标准偏差和相对误差均小于2%,以化学位移在δ:8.07处最佳定量峰计算结果的相对误差相对最小,为0.84%,测试结果最为准确可靠.

表2 不同的定量峰计算的磷酸伯氨喹含量Tab 2 Determination of primaquine phosphate content by different quantitative peaks
3 结论
本文采用qNMR法测定了磷酸伯氨喹片中磷酸伯氨喹的含量,测定无需分离过程和对照品,样品配制简单,可有效减少样品前处理对测定结果的影响.建立的qNMR法同时实现了磷酸伯氨喹片中磷酸伯氨喹的定性鉴别与定量分析,且操作简单、快速、省时,测定结果与标示值相对误差较小,说明该方法计算结果比较准确可靠.qNMR方法应用于药物定量分析应用也已成趋势,在药物质量控制包括有效成分鉴别、含量测定及杂质检测等的应用将日益广泛,是鉴别假冒、伪劣药品的有力武器,对监管我国的药品市场也有着重大的意义.
