环境中自由及结合态雌激素的酶降解转化研究进展
2019-05-09余薇薇杜邦昊张敏讷万巧玲杨硕赵晨菊
余薇薇 杜邦昊 张敏讷 万巧玲 杨硕 赵晨菊
(1. 重庆交通大学河海学院 水利水运工程教育部重点实验室,重庆 400074;2. 国家城市供水水质监测网重庆监测站,重庆 400060)
近年来,内分泌干扰物(Endocrine disrupting chemicals,EDCs)环境问题引起全球范围的密切关注和重视,是国际环境科学领域研究的前沿和热点课题之一,美国、欧盟、日本、世界卫生组织(WHO)、联合国环境规划署(UNEP)、经济合作与发展组织(OCED)等国家和组织已制定管理法规和管控措施,中国也在《水污染防治行动计划》(国发[2015]17号)中提出严格控制环境激素类化学品污染并评估风险,实施管控措施[1-3]。类固醇雌激素(Steroid estrogens,SEs)可分为天然和人工合成两大类,是雌情活性最强、最受关注的一类EDCs,在环境中被广泛检测出,主要来源于城市污水处理厂和集约化畜禽养殖场[4-7]。环境中极其微量的SEs便可强烈干扰人类和动物的内分泌系统,导致发育、生殖、神经、免疫等问题[1],长期暴露于高浓度SEs环境中还会导致鱼类雌性化、雌雄同体、植物生长损伤、微生物功能及群落多样性变化等问题[8-9],而且不同SEs间存在着复合毒性协同效应,造成的危害更大[10]。酶具有催化效率高、活化能低、反应条件温和、环境友好等特点,被誉为“绿色催化剂”,在SEs的去除中起到重要作用并广受关注[11-17]。本文总结了环境中酶对自由及结合态SEs的去除作用及其反应机理,阐明不同形态SEs的主要酶降解、转化路径,并对酶在环境中SEs去除的应用进行了展望,以期对环境中两种形态雌激素的酶降解转化的研究提供理论借鉴。
1 环境中降解转化SEs酶的基本性质
环境中天然SEs可分为自由态(Free estrogens,FEs) 和 结 合 态(Conjugated estrogen,CEs), 其结构示意如图1所示。天然SEs降解、转化过程常用的酶主要分为两大类:(1)氧化还原酶类(Oxidoreductases),通过催化氧化FEs形成聚合产物,如过氧化物酶(Peroxidases,PODs,EC 1.11.1.X)和漆酶(Laccase,Lac,EC 1.10.3.2)[18-21];(2)水解酶类(Hydrolases),通过解离结合基团将CEs水解为相对应的FEs,如芳基硫酸酯酶(Arylsulfatase,ArySTS,EC 3.1.6.1)和β-葡糖苷酸酶(β-glucuronidase,GUSB,EC 3.2.1.31)[22-25]。典型 SEs降解、转化酶的物理化学性质及三维结构,见表1和图2。除以上几种酶外,还有一些细菌产生的酶对SEs具有降解、转化作用,如亚硝化单胞菌属(Nitrosomonassp.)可产生单加氧酶(Monooxygenase)、诺卡氏菌属(Nocardiasp.)及不动杆菌属(Acinetobactersp.)可产生双加氧酶(Dioxygenase)、红球菌属(Rhodococcussp.)及鞘氨醇单胞菌属(Sphingomonassp.)可产生单加氧酶、双加氧酶、羟基类固醇脱氢酶(Hydroxysteroid dehydrogenase,HSD)等[26-27]。
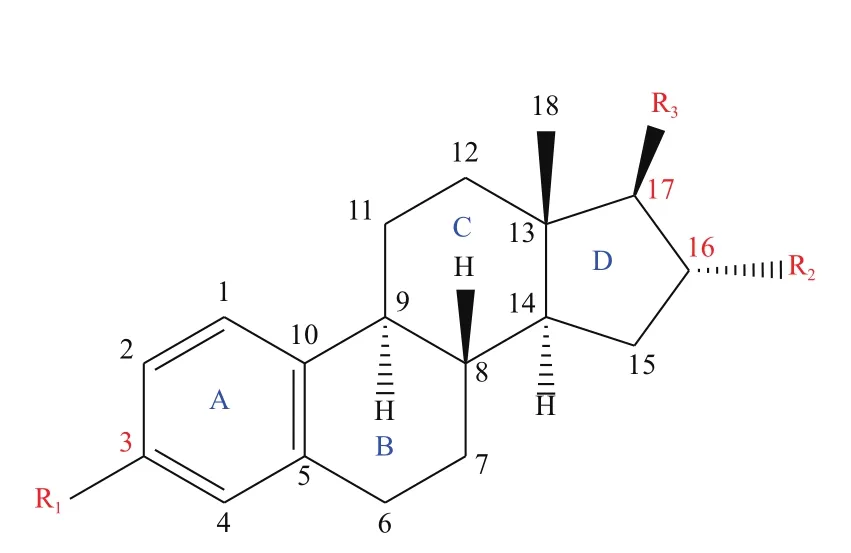
图1 天然SEs结构示意图
2 氧化还原酶对FEs的降解转化作用
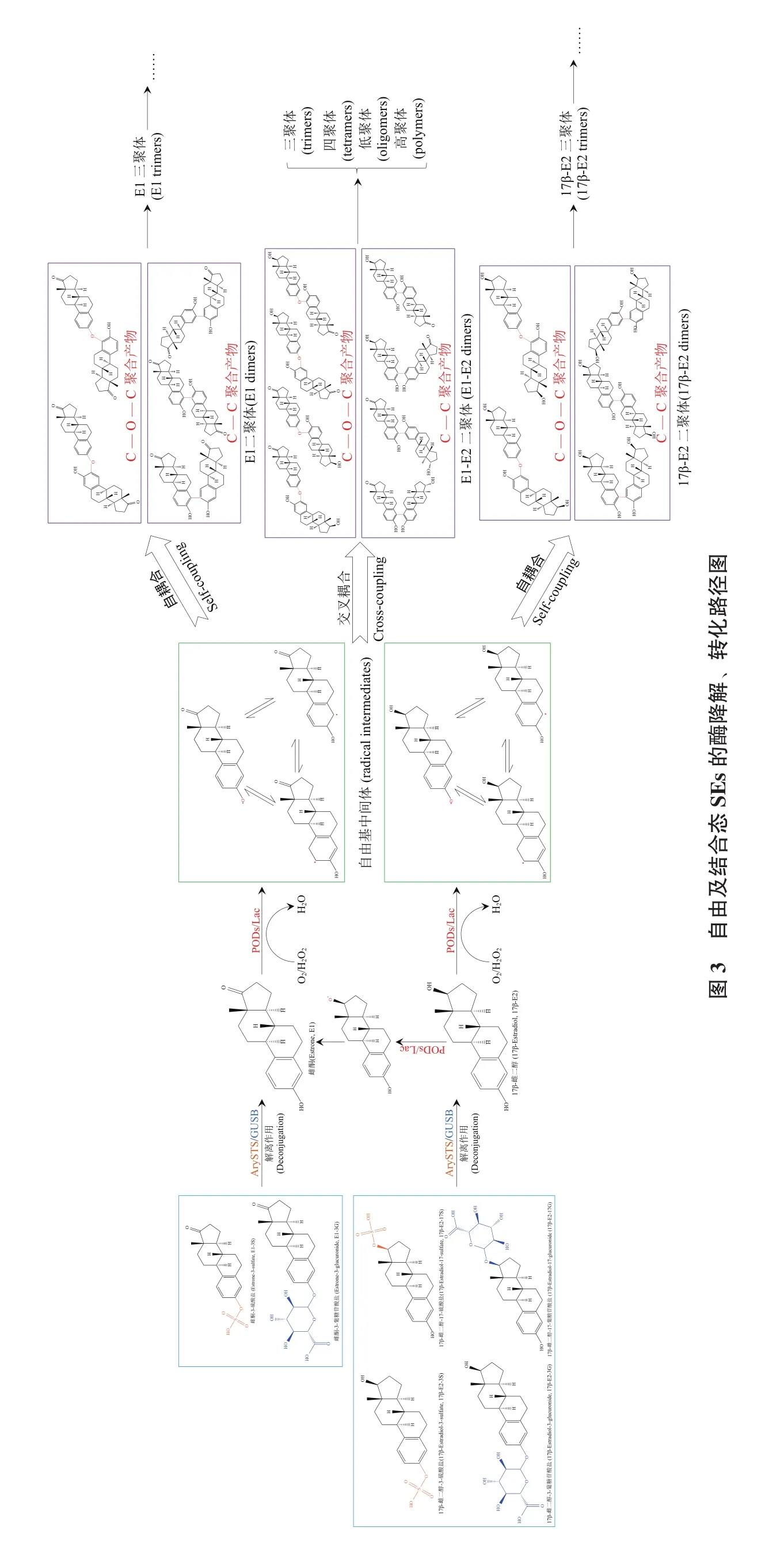
FEs主要包括雌酮(Estrone,E1)、17α-雌二醇(17α-Estradiol,17α-E2)、17β-雌二醇(17β-Estradiol,17β-E2)、雌三醇(Estriol,E3)。氧化还原酶去除FEs的主要作用机制为氧化耦合反应,催化FEs A环酚羟基形成高氧化潜能、高活性的C2、C4位自由基与C3位苯氧自由基等自由基中间体(图3),通过C-O-C、C-C共价键自耦合、相互耦合形成二聚体,同时受到溶剂类型、pH、A环取代位置等影响[14,18,21,31-32]。FEs A 环 C3 位氧原子的电荷密度较高而自旋密度较低,因此与C-O-C键相比,在动力学上更有利于形成C-C键聚合产物[33]。二聚体可继续形成自由基,作为酶的底物继续氧化耦合生成三聚体、低聚体,甚至高聚体,但聚合物的溶解度随相对分子质量增加急剧降低,难以进一步耦合形成高分子量的聚合物[32,34]。氧化还原酶催化FEs生成的聚合产物溶解度较低,在污水处理过程中易于通过吸附、过滤、沉淀等作用被去除。氧化还原酶的催化能力通常与氧化还原电位(表1)和糖基化程度相关,高氧化还原电位有利于酶对SEs的催化氧化,在一定范围内,酶的稳定性随糖基化程度的增加而提高,其中LiP的糖基化程度较高,为20%-30%,高于HRP(18%-22%)、Lac(10%-20%)和MnP(5%-15%)[35-36]。PODs和 Lac催化机理的主要区别在于电子受体,通常PODs以H2O2作为电子受体催化其活性,同时还需要辅酶因子的参与,而Lac以空气或水中的氧分子作为电子受体,不需额外添加氧化剂[17,34,37-39]。FEs在 Lac 催化体系中的去除速率低于PODs,但PODs不稳定,在复杂基质及过量H2O2条件下易于失活,而Lac的稳定性更强[34]。另外,与A环C3位相结合的CEs(如E1-3S、17β-E2-3S、E1-3G、17β-E2-3G)受到硫酸盐或葡糖苷酸盐基团的保护,不易被氧化还原酶催化氧化形成自由基[22]。不同基质中氧化还原酶对FEs的去除效率如表2所示。
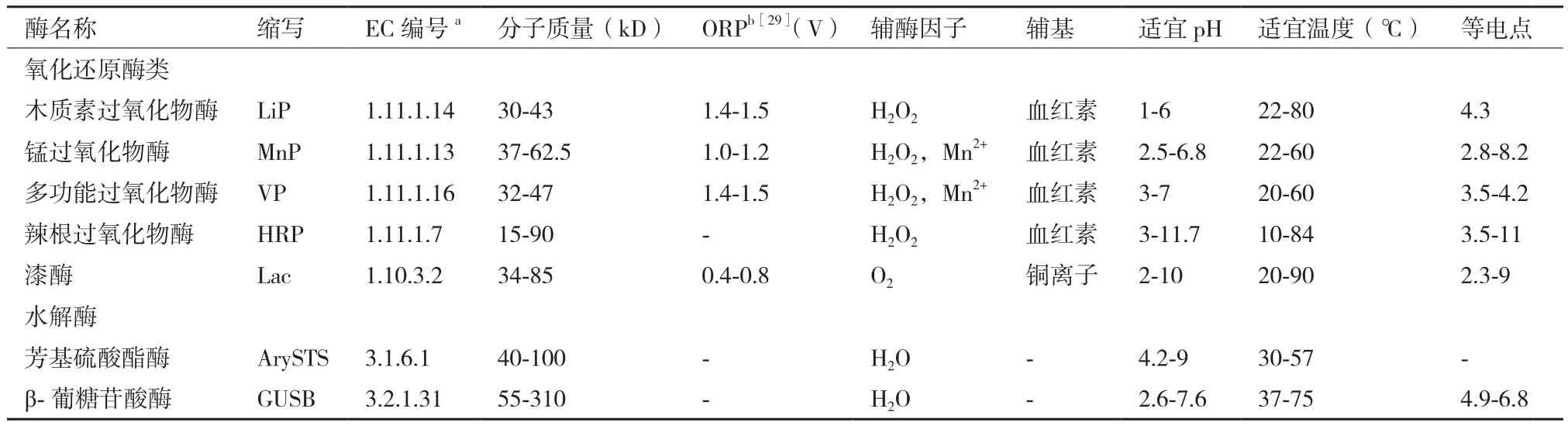
表1 典型SEs降解、转化酶的主要物理化学性质[28]
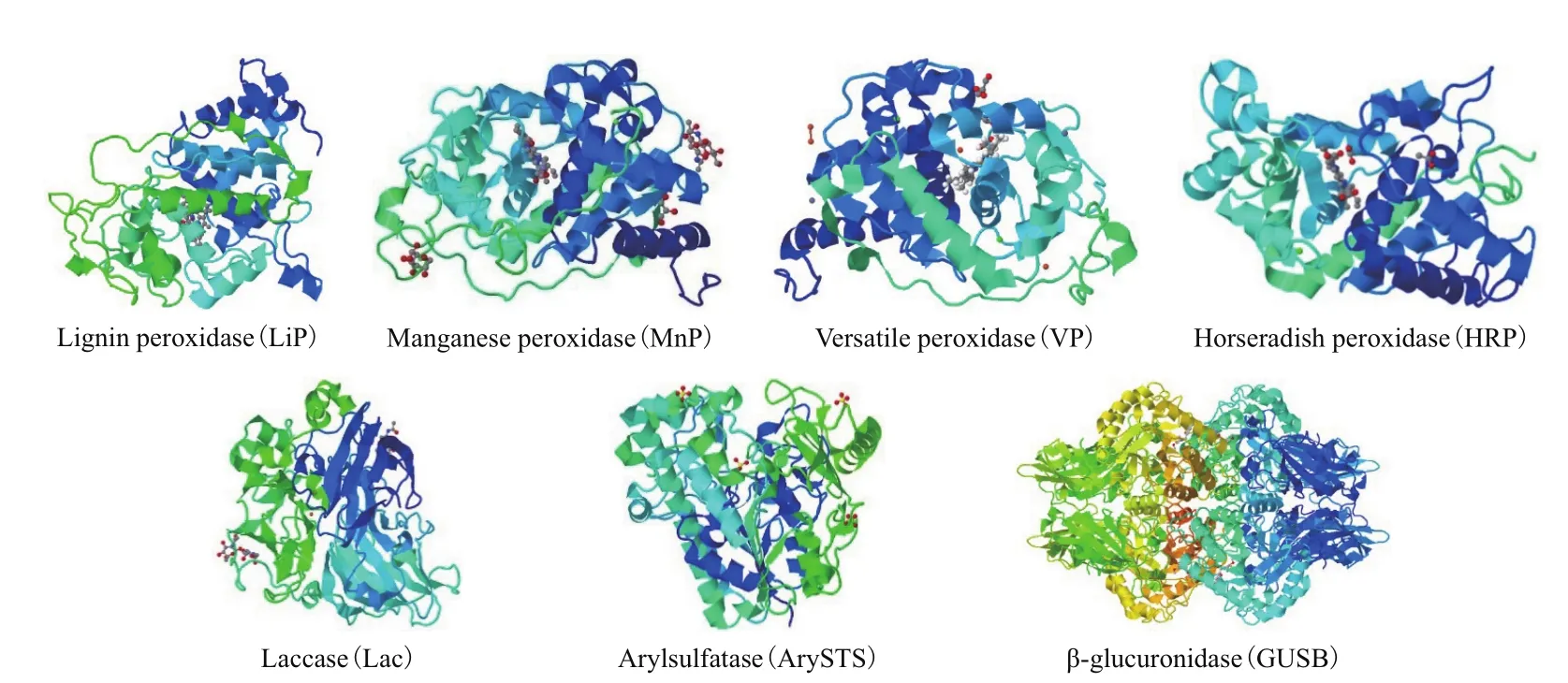
图2 典型SEs降解、转化酶三维结构示意图[30]
2.1 过氧化物酶的降解作用
PODs在自然界中分布广泛,可由动植物、真菌、细菌产生,根据是否含血红素分为血红素(Heme-peroxidases)及非血红素过氧化物酶(Nonheme peroxidases)[51-52]。在 FEs的 催 化氧化中应用较多的PODs主要为血红素过氧化物酶,包括两大类:(1)真菌分泌的胞外木质素降解酶(Lignin modifying enzymes,LMEs),如 LiP、MnP、VP。(2)植物产生的HRP[51]。PODs的催化机理为:血红素蛋白被H2O2或小分子过氧化物氧化为化合物I(卟啉π阳离子自由基氧铁基团,FeIV=O·+),随后化合物I从底物得到一个电子还原为化合物Ⅱ(氧铁基团,FeIV=O),再氧化底物得到一个电子将酶还原为基态FeIII,其中最后一步为限速步骤[13,53],FEs被催化氧化生成自由基后通过耦合反应生成聚合产物。

表2 氧化还原酶对不同基质中FEs的去除
2.1.1 木质素降解酶 LiP在酸性条件下催化去除FEs效果较好,Wang等[41]实验结果表明,在pH3,0.2 mol/L H2O2条件下,120 U/L LiP对2.7 mg/L E1和17β-E2的去除率为60%-90%。Mao等[19]实验表明在 pH4.6,0.05 mmol/L H2O2条件下,2.5 mg/L 17β-E2可被20 U/L LiP完全去除,主要产物为17β-E2低聚物,少量转化为中间产物E1。藜芦醇作为白腐真菌(White-rot fungi,WRF)的次生代谢产物以及与LiP共生的活性底物,具有较强的氧化能力,在LiP催化FEs过程中可被氧化成阳离子自由基,保护LiP避免失活并增强其活性,且对失活的LiP有一定的恢复作用[19,32,40]。无藜芦醇时,初始浓度为5 mg/L的17β-E2反应60 min后去除率仅为40%,而添加藜芦醇能够提高LiP对FEs的催化去除效率,反应 20 min 便能将 5 mg/L 17β-E2 完全去除[19]。
MnP是一类以多种形式存在的糖基化血红素过氧化物酶,其结合位点的结构与其他PODs有所不同,通过将Mn2+氧化为Mn3+完成催化氧化[54]。Tamagawa等[44]试验结果表明,经600 U/L MnP处理1 h后,0.01 mmol/L E1被完全去除,2 h后雌激素活性得以完全去除。
VP是一种混合酶,结合了LiP和MnP两种酶的底物特异性,在H2O2、Mn2+以及底物的共同作用下,将Mn2+氧化为Mn3+,催化氧化FEs形成聚合产物[21]。Eibes等[42]考察了 VP 对 E1 和 17β-E2 的去除,结果表明在低酶活性(10 U/L)条件下,反应25 min内两种化合物能够被完全去除。Taboada-Puig等[21,43]实验结果表明,在最优条件下,VP可去除污水中高浓度(1.3-8.8 mg/L)及环境浓度(1.2-6.1 μg/L)的E1和17β-E2,并形成二聚体、三聚体和羟基雌酮(Hydroxyestrone,OH-E1)等产物。
2.1.2 辣根过氧化物酶 HRP是一种含铁卟啉辅基的PODs,含有两个不同的金属中心:血红素和两个钙原子,主要由植物分泌,在辣根(Horseradish)中含量最高,并因此而得名[35,55]。Auriol等[18,56]研究发现在pH为7,温度为25℃条件下,被H2O2激活后,32 U/L HRP在1 h内可去除人工污水中97%的100 ng/L E1、17β-E2或E3,反应5 h后去除率为99%;但在实际污水中,去除相同浓度的FEs需添加8 000-10 000 U/L HRP,说明复杂污水基质成分使得HRP氧化能力受到抑制,从而显著影响了FEs的氧化去除。
值得注意的是,H2O2作为PODs的电子受体,同时又是这几种酶的抑制剂,过量H2O2会使反应过程中过渡态化合物形成无反应活性的化合物III(铁超氧化物,FeIII=O2·-),称之为“自杀性失活”(Suicide inactivation)[13,57]。而作为一种混合酶,VP 的“自杀性失活”更加复杂,由于其存在3个氧化位点及多个催化产物,对H2O2浓度最敏感[57]。有研究表明,添加稳定剂可保护酶活性避免失活,如聚乙二醇(Polyethylene glycol,PEG)、聚乙烯醇(Polyvinyl alcohol,PVA)、聚乙烯(Polythene,PE)、明胶(Gelatin)、多糖(Polysaccharide)等[13,36]。
2.2 漆酶的催化氧化作用
Lac是一类多酚氧化酶,自然界中Lac可由高等植物、昆虫、细菌、真菌产生,其中真菌漆酶(Fungal Laccase)的底物特异性低,氧化能力强,应用最为广泛[14,37,51-52]。Lac 催化活性中心通常含有 4 个铜离子,三环的铜簇位点以空气或水中的氧分子作为电子受体,形成自由基后,T1铜中心参与FEs单电子氧化,将O2还原为水,生成C-O-C、C-C聚合产物[11,55,58]。FEs在 A 环 C3 位含有酚羟基,能够被Lac催化产生苯氧自由基,通过氧化耦合反应形成二聚体、三聚体,有研究表明Lac还可将17β-E2氧化为 E1[20,39,50,59],而与之相反,Singh 等[48]在反应产物中未检测出E1和E3。
Lloret等[39]研究发现,在水相反应体系中,pH4时Lac的酶活性最高,但稳定性较低,而其酶活性在pH7时较低,但稳定性高,同时可观察到反应初期Lac在pH4时有显著失活,而在pH7条件下,6 h内酶活性较稳定。Singh等[55]实验研究表明,在2 500 U/L Lac条件下,在水相体系中反应5 h后0.2 mg/L 17β-E2去除率为93%,而在土壤中去除率为87.1%。土壤含水率对Lac处理效果也有影响,经24 h 10 U/g Lac处理后,非饱和土壤中0.2 mg/g 17β-E2去除率仅为32.1%,而在饱和土壤中去除率达到62.6%,这可能由于Lac酶分子在水相中有利于与17β-E2接触,从而去除效果增强[48]。在pH为7,温度为25℃条件下,20 000 U/L Lac在1 h内可完全去除人工及实际污水中100 ng/L E1、17β-E2或E3,实验结果表明其催化FEs耦合反应受污水基质成分的影响较小[46]。有研究表明在相同浓度下,Lac催化氧化E1的反应速率仅为17β-E2的60%,其区别在于二者在C17位与Lac的亲和性[22]。Lac的稳定性在水相和土壤体系中有较大差异,虽然在水相(0.46 h-1)中其失活速率比在土壤(0.003 1 h-1)中快很多,但水相中Lac对17β-E2的去除速率比在土壤中高100多倍[48],Lac在土壤中的活性持续时间较长、稳定性强的特点弥补了其反应速率慢的缺点。
漆酶介导反应体系(Laccase mediator system,LMS)指Lac在介体和氧分子存在时进行催化氧化的反应体系,介体的氧化还原电位较高,可分为天然及人工合成介体,如紫脲酸(Violuric acid,VA)、丁香醛(Syringaldehyde,SA)、1-羟基苯并三氮唑(1-hydroxy-benzotriazole,HBT)、2,2-联氮 -二(3-乙基-苯并噻唑-6-磺酸)二铵盐(2,2-azinobis(3-ethylbenzothiazoline-6-sulfnoic acid),ABTS)等[45-46,60-62]。介体在酶和目标化合物间起到“电子穿梭”的作用,作用机制主要包括氢自由基转移、电子转移以及离子氧化,添加介体可提高FEs的氧化程度,从而增强 Lac 催化去除 FEs反应[17,61,63]。Auriol等[46]实验结果表明,当Lac酶活性低于20 000 U/L时添加HBT可提高17β-E2的去除效率,而当酶活性高于20 000 U/L时,两种条件下17β-E2的去除率没有明显差异。Sei等[64]实验结果表明,在无介体条件下,300 U/L Lac反应6-12 h后可完全去除20 mg/L 17β-E2和E3,24 h后E1的去除率为90%,添加介体ABTS或HBT后E1仅在3 h内可被Lac完全去除。Lloret等[62]实验表明,不添加介体条件下,经Lac处理24 h后17β-E2得以完全去除,而E1不易去除,在Lac-VA、Lac-HBT和Lac-SA体系中E1的去除率分别为100%、88%和74%。在以后的研究中,应探索成本低、性质稳定、毒性低及水溶性好的介体。
2.3 有机质对氧化还原酶催化FEs的影响
土壤与水体环境中富含有机质,有研究表明高浓度有机质可显著降低FEs的酶催化去除速率,这是由于有机质含有大量酚类官能团,形成自由基后可与FEs及其自由基相互耦合,形成交叉耦合化合物,从而抑制FEs自耦合产物的形成[33-34,65-66]。Mao等[32]试验证实,藜芦醇对LiP催化能力的增强会受到有机质的抑制。当有机质和藜芦醇同时存在时,二者在LiP表面活性位点产生竞争,导致17β-E2的去除率有所降低,这是由于有机质和17β-E2的交叉耦合反应抑制了藜芦醇对LiP去除17β-E2的增强作用[23,40]。Huang 等[65]研究了有机质对 HRP去除17β-E2的影响,发现有机质不仅抑制17β-E2的去除,还会影响17β-E2氧化自耦合产物二聚体、三聚体的形成。Sun等[66]研究发现有机质可与Lac表面活性位点竞争或与17β-E2结合,从而抑制17β-E2的酶催化氧化去除,且其抑制率随有机质浓度增加而升高,有机质浓度为50 mg/L时,Lac对17β-E2的去除率不足5%。夏青[55]通过试验研究表明,有机质对HRP去除E1、17β-E2的抑制作用并不明显,去除率仍高于80%,而在Lac催化体系中,有机质对E1、17β-E2的去除有抑制作用,说明有机质对HRP活性的影响较小。
3 水解酶对CEs的降解转化作用
FEs羟基可与硫酸盐或葡糖苷酸盐通过酯化作用形成CEs,其中单羟基化合物E1的结合态为单一形式,多羟基化合物E2、E3则可与一个或多个结合基团形成硫酸盐(CSEs)、葡糖苷酸盐(CGEs)、硫酸盐-葡糖苷酸盐(CS-GEs)形式的CEs。与FEs相比,CEs的水溶性、迁移性高,而吸附性较低。研究普遍认为CEs无生物毒性,但其解离后又重新生成雌情活性高的FEs,潜在的生态风险不应被忽视。CEs结合基团的解离主要为ArySTS和GUSB的作用,其活性与温度及微生物丰度密切相关,酶催化条件下CEs的解离在热力学上不可逆[16]。CGEs可被GUSB解离为相对应的FEs,ArySTS通过水解硫酯键将CSEs酶解为其对应的FEs[25,67]。研究表明,GUSB和ArySTS活性与有机质含量、土壤黏粒显著相关[23,68-69]。目前有关CEs酶降解、转化的研究多集中在E1及E2的结合态,而有关E3结合态形式酶解的研究相对较少。
3.1 芳基硫酸酯酶的水解作用
ArySTS可由微生物、动植物分泌,在污水及活性污泥中活性和含量水平较高[70-71]。土壤中ArySTS主要以吸附态存在于腐殖质、黏土矿物,稳定性增加且不易失活,而很少以游离态形式存在[68],Bai等[72]研究也表明ArySTS仅在土壤吸附相中保持活性,而在水相中几乎没有活性。水体中ArySTS主要来自底泥微生物的分泌,也可通过径流、合流制污水管网溢流等方式携带产生ArySTS的微生物进入水体[25,70]。然而,由于环境中ArySTS活性与丰富度水平相对较低,CSEs硫酯键的解离作用通常较弱[73]。Liu等[74]的实验结果表明,在1 mL pH5的缓冲液,55℃条件下,添加50 μL ArySTS反应3 h后,E1-3S和E3-3S的解离效率超过75%,而17β-E2-3S的解离效率低于60%。Bai等[72,75]实验表明,ArySTS对17β-E2-17S的解离作用仅为A环羟基化作用的1/10,主要产物为 OH-17β-E2-17S 和 diOH-17β-E2-17S。Ma和Yates[76-77]也发现农田土壤、河水及沉积物中17β-E2-3S主要通过C17位羟基的氧化生成初级代谢产物E1-3S,而酶的解离作用次之。另外,氧化还原条件也会影响ArySTS的活性,Zheng等[78]测定了奶牛养殖场污水17α-E2-3S的转化产物,结果表明,在有氧条件下主要通过D环C17位的羟基氧化为酮基,从而生成E1-3S,而在厌氧条件下A环C3位硫酯键的酶解占据主导代谢地位。
值得注意的是,芳基磺基转移酶(Arylsulfotransferase,ArySULT,EC 2.8.2.1)与ArySTS的作用则相反, 能 够 将 FEs重 新 转 化 为 CSEs[67]。Goeppert等[67,79]研究了土壤中17β-E2的转化路径,结果表明17β-E2可先降解为E1,随后在ArySULT的作用下转化为E1-3S。
3.2 β-葡糖苷酸酶的水解作用
GUSB属糖苷类水解酶,细菌、真菌及高等动植物均可产生,广泛存在于污水、活性污泥、土壤、沉积物中[23]。在河水及沉积物中,Ma和Yates[76-77]观察到17β-E2-3G主要通过C3位结合基团的解离生成17β-E2,而在农田土壤中主要为C17位羟基氧化作用生成E1-3G,这可能由于土壤中GUSB含量水平不足所致。Kumar等[80]实验结果表明,在河水中反应5 d后,E1-3G完全解离为E1,而仅有64%的17β-E2-3G解离为17β-E2及随后的转化产物E1,这可能由于GUSB对两种CGEs的解离存在差异。
粪大肠菌群,例如埃希氏大肠杆菌(Escherichia coli,E. coli)可合成GUSB和ArySTS,从蜗牛(Helix pomatia,H. pomatia)中提取的GUSB-ArySTS混合酶也广泛应用于 CEs的酶解及检测[71,74]。E. coli合成的ArySTS较少且活性、亲和性相对较弱,H.pomatia中提取的GUSB-ArySTS混合酶中ArySTS活性也相对较低[23,81],因此与 CGEs相比,CSEs在环境中的稳定性和持久性更强[80,82]。Ben 等[82]实验表明,10 μmol/U酶抑制剂STX 64和D-葡糖二酸-1,4-内酯可分别有效抑制污水中ArySTS和GUSB活性,从而阻碍CEs的解离。结合基团(硫酸盐或葡糖苷酸盐)及位置(A和/或D环)的差异会影响CEs的酶降解、转化过程。D’Ascenzo等[83]的实验结果表明,A环(E1-3G、17β-E2-3G、E3-3G)CGEs比D环(17β-E2-17G、E3-16G)CGEs易于解离,不过一旦反应启动,一天内均可完全解离为相对应的FEs;而对于CSEs,E1-3S、17β-E2-3S和E3-3S完全解离需6-8 d,说明污水中ArySTS活性相比于GUSB弱得多。Liu等[16]比较了5种CEs的解离速率,并划分为低(E1-3S)、中等(E1-3G、17β-E2-3S和E3-3G)和高(E3-16G)解离速率。Gomes等[84]通过活性污泥批试验表明,CEs(E1-3S、E1-3G和E3-16G)在微生物分泌的酶作用下可解离为FEs,而灭菌条件下未检测到FEs,解离优先顺序为E1-3G>E3-16G>E1-3S,其影响因素为:结合基团>基团位置>SEs类型。
水解酶通过解离硫酸盐或葡糖苷酸盐结合基团将CEs水解为相对应的FEs,氧化还原酶可催化FEs形成C2、C4位自由基及C3位苯氧自由基中间体,通过C-O-C、C-C共价键自耦合、交叉耦合形成二聚体、三聚体,甚至高聚体。环境中自由及结合态SEs的酶降解、转化路径如图3所示。
4 展望
酶对环境中自由及结合态SEs的去除的应用受到诸多因素限制,对今后酶在SEs降解转化的规模化应用、机理研究、联合应用、固定化酶、酶工程等方面进行了展望。
酶催化去除SEs的研究大多处于小试、中试试验阶段,在污水、污染土壤修复中的应用相对较少[31,43,47,49,52],由于理论研究与实际应用存在差异,评估酶在实际应用中的可行性十分重要。污水中各种干扰因素会对酶的催化氧化效果产生影响,如pH、温度、盐度、溶解氧、重金属、抑制物质等[36]。应进一步研究酶处理含SEs污水的工作机理和环境因素的影响,将理论应用于实际工业化处理中。酶膜反应器(Enzymatic membrane reactor,EMR)可应用于污水中有机污染物连续处理[14,38,85],目前已有研究在小试阶段应用EMR处理FEs,去除效率可达到 80%-100%[20,43,60],常用的膜材料有醋酸纤维素(Cellulose acetate,CA)、硝酸纤维素(Nitrocellulose,NC)、聚四氟乙烯(Polytetrafluoroethylene,PTFE)、聚丙烯腈(Polyacrylonitrile,PAN)、聚醚砜(Polyethersulfone,PES)、聚酰胺(Polyamide,PA)等[85]。在规模化应用时,需要解决酶失活以及膜的持续性能问题,同时,EMR在SEs特别是CEs去除方面的应用仍需进一步开发和研究。酶催化应用于SEs污染土壤修复不会产生二次污染,然而不同土壤类型、气候条件、微生物群落等均会对酶修复SEs污染土壤产生影响,实现场地修复仍需要长期系统地研究。
在酶处理SEs过程中伴随着中间产物的产生,且有些产物仍具有雌情活性,而大多数研究没有检测反应后总雌情活性,以及对降解、转化产物进行鉴定。随着高分辨质谱(HRMS)、核磁共振(NMR)等分析检测技术的发展[58,86],应在反应中间产物的鉴定,自由基的生成,C-O-C、C-C键聚合产物优先顺序及比例等方面展开研究,进一步阐明SEs的酶催化反应机理,比较不同类型的酶对SEs的去除效率,深入分析SEs的降解、转化产物及路径。
WRF(如Trametes versicolor、Phanerochaete chrysosporium)可分泌Lac、LiP、MnP,且不同种类的菌分泌的酶类型存在差异,利用WRF分泌的胞外酶体系催化氧化FEs,可作为规模化应用发展的方向[51,59]。通常污水、污泥、土壤中存在着多种类型的自由态和结合态SEs,是十分复杂的混合体系,而单一种类的酶很难同时去除不同形态的SEs。在GUSB-Lac联用酶体系处理污水中不同形态SEs(E1、17β-E2、17β-E2-3G)的试验中,Tanaka等[22]发现几种化合物均能够得到有效去除。水解酶与氧化还原酶联用同时处理复杂基质中的FEs和CEs(CSEs和CGEs),或产生这些酶的微生物的联合使用(如WRF和E. coli)可作为今后研究的重要课题。
溶解态的游离酶在污水中存在易变性失活、易流失、难以回收等问题,从而导致处理成本过高,限制了其大规模应用。酶固定化技术可以提高酶的稳定性,实现重复利用,并在SEs的去除中得以应用[54,87-89],主要分为物理法(如吸附)和化学法(如包埋、微胶囊、交联、共价结合),且各有优缺点,载体材料及固定化技术的选择取决于酶的类型和催化过程[54,90]。探寻经济、高效、生物相容且环境友好的固定化材料,降低酶活性损失并提高稳定性是未来发展的方向。
自然条件下微生物产生酶的量非常少,提高酶产量和催化效率,降低应用成本等是需要解决的主要难题,可通过酶工程(如纳米酶、修饰酶和超酶)、基因编辑、DNA重组等技术提高酶产量、稳定性及活性、催化效率及降低酶的别构调节[51-52,91]。
