乳酸脱氢酶与葡萄糖脱氢酶偶联催化合成D-苯基乳酸
2019-05-07罗希杨泽锋臧瑜李宁王鑫情付永前
罗希,杨泽锋,臧瑜,李宁,王鑫情,付永前
(台州学院 生物质资源研究所,浙江 台州,318000)
D-苯基乳酸(D-phenyllactic acid,D-PLA),是一种由乳酸菌等微生物产生的天然抑菌物质,安全无毒,具有抗菌谱广、水溶性好、稳定性高、有效pH范围宽等特点,在食品工业中有着广阔的应用前景[1-4]。D-PLA用于化妆品中,不仅能有效防止微生物污染引起的感染,而且具有去皱、亮肤的功效[5]。D-PLA可作为动物饲料添加剂取代抗生素,减少家畜大肠内大肠杆菌的数量,提高家畜体内免疫相关类血细胞的数量,促进个体生长[6-7]。D-PLA可作为单体生产高分子聚合物聚苯乳酸,具有高水平的紫外吸收特性[8-9]。除此之外,D-PLA是丹参素衍生物,临床上可治疗冠心病[10-11],也可作为化学原料合成非蛋白氨基酸、抗HIV试剂和降血糖剂等[12-13]。D-PLA在食品、化妆品、农业、医药、化学工业等领域广泛应用且具有巨大的市场需求。
乳酸脱氢酶(lactate dehydrogenase, LDH)是D-PLA酶法合成的关键酶,可催化苯丙酮酸钠(sodium phenylpyruvate, PPA)还原生成D-PLA。大肠杆菌(E.coli) BL21是外源蛋白表达通用宿主,易于操作和控制,近年来多株LDH重组大肠杆菌被构建,以PPA为底物不对称还原合成D-PLA,但LDH基因来源较少,主要集中在乳杆菌属[14-16]Wickerhamiafluorescens[17],Pediococcusacidilactici[18],Pediococcuspentosaceus[19]等少数微生物菌种。ZHU等对LactobacilluspentosusD-乳酸脱氢酶进行定点突变,以E.colipET28a-ldhY52V为催化剂在水-正辛烷两相介质中催化合成D-PLA,时空产率达到310.08 g/(L·d),为目前已知的最高生产强度[20]。除对已有LDH进行分子改造提高催化效率外,新酶的发现也是生物催化领域的重要课题。LDH属于氧化还原酶家族,其催化活性依赖于烟酰胺辅酶[nicotinamide adenine nucleotide (phosphate), NAD(P)H],辅酶将氢和电子传递给底物羰基,使底物还原[21]。而烟酰胺类辅酶价格昂贵,稳定性低,难以重复利用,增加了反应成本,极大地制约了乳酸脱氢酶的工业化应用。因此,烟酰胺辅酶的再生效率在工业生物催化中起着至关重要的作用[22-23]。为了克服辅酶的供应问题,葡萄糖脱氢酶(glucose dehydrogenase, GDH)常被用于辅酶的再生[24-26]。本实验挖掘了1株新型LDH,与GDH偶联,在实现辅酶再生的同时催化合成D-PLA(图1)。

图1 双酶耦联不对称还原PPA合成D-PLA
Fig.1 Asymmetric reduction of PPA to D-PLA bycoupled LDH and GDH
1 材料与方法
1.1 材料与试剂
TaqDNA聚合酶、PfuDNA聚合酶、限制性内切酶、T4 DNA连接酶:Takara;胰蛋白胨和酵母粉:Oxoid;DNA胶回收试剂盒、质粒小量提取试剂盒、PCR产物清洁试剂盒、基因组提取试剂盒、限制性内切酶、苯丙酮酸钠、D-苯基乳酸、辅酶NADH和pUCm-T质粒:生工生物科技有限公司;菌种Lactobacillusrossiae、E.coliBL21 (DE3)、E.coliBL21 (DE3)/pET-28a-esgdh、质粒pET28a(+):本实验室保藏;色谱纯乙腈、甲醇、甲酸、三氟乙酸:百灵威科技有限公司;其他化学试剂均为市售分析纯。
1.2 仪器与设备
CHA-SA气浴恒温振荡器,上海江星;LC-20A高效液相色谱仪,岛津;HICO21小型高速离心机,上海生工;Avanti J-E高速冷冻离心机,Beckman;BPMJ-70F恒温培养箱,上海一恒。
1.3 乳酸脱氢酶基因克隆
根据Lactobacillusrossiae乳酸脱氢酶(LDH,GenBank: AZFF01000004.1)核苷酸序列设计引物(LDH-F: CCATGGGCATGGAGGTGTCTGCATTGA; LDH-R: CTCGAGCTAGTTAAAGGCCACAACA T)。以L.rossiae基因组DNA为模板进行PCR,克隆基因片段。PCR反应体系(总体积100 μL):10×PfuDNA Polymerase Buffer 10 μL(Mg2+),引物LDH-F和LDH-R各1 μL (50 μmol/L),dNTP mixture 1 μL(10 mmol/L),基因组DNA 1 μL,PfuDNA Polymerase 1 μL,去离子水85 μL。PCR程序为:94 ℃预变性5 min,经过35个循环(94 ℃ 0.5 min,56 ℃ 0.5 min,72 ℃ 1.5 min),72 ℃延伸 10 min,4 ℃保温。琼脂糖凝胶电泳,检测是否获得目的条带并胶回收目的基因。
1.4 乳酸脱氢酶重组基因工程菌的构建
目的基因与pUCm-T质粒连接,构建的重组T质粒和pET28a(+)质粒用相同的限制性内切酶(NcoI/XhoI)切割,产生的黏性末端用T4 DNA连接酶连接,构建重组表达质粒pET28a-ldh,转化BL21(DE3)大肠杆菌感受态,构建重组菌E.coliBL21 (DE3)/pET28a-ldh。将乳酸脱氢酶重组菌接种至含30 μg/mL卡那霉素的LB液体培养基中,37 ℃、180 r/min培养至发酵液OD600值为0.8,加入终质量浓度为5 g/L的乳糖,28 ℃,150 r/min诱导12 h,离心收集菌体,磷酸缓冲液(100 mmol/L,pH 7.0)悬浮,超声破碎并冷冻离心,取上清,SDS-PAGE凝胶电泳分析。
1.5 乳酸脱氢酶重组菌诱导表达条件优化
接种E.coli/pET28a-ldh至含有30 μg/mL卡那霉素的LB液体培养基,37 ℃培养10 h,再以体积分数4%接种到含有30 μg/mL卡那霉素的LB液体培养基中,37 ℃培养至菌液OD600值为0.8,加入乳糖,在28 ℃,150 r/min下诱导,分别对诱导剂乳糖的质量浓度2、4、6、8、10、12 g/L和诱导时间2、4、6、8、10 h进行优化。离心收集菌体、测生物量,计算比酶活。
生物量测定:取1.0 mL发酵液,离心收集菌体,并用生理盐水洗涤2次后,置于80 ℃烘箱中干燥至恒重并称量。
重组乳酸脱氢酶活力测定:诱导表达后的菌体细胞用磷酸缓冲液(100 mmol/L,pH 7.0)悬浮,菌体质量浓度以干菌体计为20 g/L,超声破碎并离心,取上清作为粗酶液。酶活检测体系由上述粗酶液、0.5 mmol/L NADH和0.5 mmol/L苯丙酮酸钠构成,总体积为200 μL,置于30 ℃、150 r/min下反应10 min,液相测定苯基乳酸含量,并计算比酶活。
酶活单位(U)定义为:在30 ℃、pH值7.0条件下,1 min还原生成1 μmolD- PLA所需要的酶量定义为1 U。比酶活定义:1 g干菌体所具有的活力单位数。
1.6 乳酸脱氢酶/葡萄糖脱氢酶偶联催化体系的构建与反应条件的优化
E.coli/pET28a-ldh和E.coli(DE3)/pET28a-esgdh两菌种分别在优化后的条件下进行诱导培养,收集的菌体按一定干重比混合后,用100 mmol/L磷酸缓冲液悬浮,菌体总质量浓度以干菌体计,为20 g/L,超声破碎。取破碎液10 mL加入10 g/L PPA和一定浓度葡萄糖,在200 r/min下反应30 min。分别对催化反应pH(6.0、6.5、7.0、7.5、8.0),温度(27、30、35、40、45 ℃),LDH和葡萄糖脱氢酶重组菌质量比(1、2、3、4、5),葡萄糖与PPA摩尔浓度之比(0.5、1、1.5、2、3、4、5)进行优化。反应生成PLA浓度和对映体过量值(enantiomeric excess,ee)用高效液相色谱检测。
1.7 双酶耦联催化反应时间进程
取上述混合破碎液50 mL,分别加入10、20、30 g/L PPA及2倍摩尔浓度的葡萄糖至100 mL圆底烧瓶中。在30 ℃、磁力搅拌转速为300 r/min下反应,流加1 mol/L Na2CO3溶液使反应液pH维持在7.0。每20 min取样,液相检测PLA浓度和ee。
1.8 液相检测方法
PLA的浓度采用Hypersil ODS C18分析柱(250 mm×4.6 mm, 5 μm)检测,流动相为V(乙腈)∶V(0.1%甲酸水溶液)=1∶ 4,流速1.0 mL/min,进样量20 μL,检测波长210 nm,柱温40 ℃。
ee采用Chiralcel OJ-RH分析柱(4.6 mm×150 mm, 5 μm)检测,流动相为V(乙腈)∶V(甲醇)∶V(三氟乙酸)∶V(水)=50∶50∶1.5∶898.5,流速0.6 mL/min,进样量20 μL,检测波长210 nm,柱温40 ℃。
1.9 ee计算方法
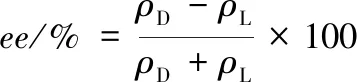
(1)
式中:ρD和ρL分别为D-PLA和L-PLA的质量浓度。
2 结果与分析
2.1 乳酸脱氢酶重组工程菌的构建
PCR扩增产物进行琼脂糖凝胶电泳,在约1 000 bp处显示了明亮条带(图2-a),与目标基因长度(1 014 bp)一致。将扩增获得的基因片段插入pET-28a(+),构建重组表达载体pET-28a-ldh转化大肠杆菌BL21 (DE3)感受态细胞,经测序确认,成功构建了重组大肠杆菌E.coli/pET28a-ldh。对诱导后的LDH重组菌破碎上清进行SDS-PAGE凝胶电泳,以无重组载体的E.coliBL21 (DE3)作空白对照,如图2-b所示,获得约40 kDa条带,与LDH预测大小一致(37.8 kDa),表明LDH重组菌成功表达。
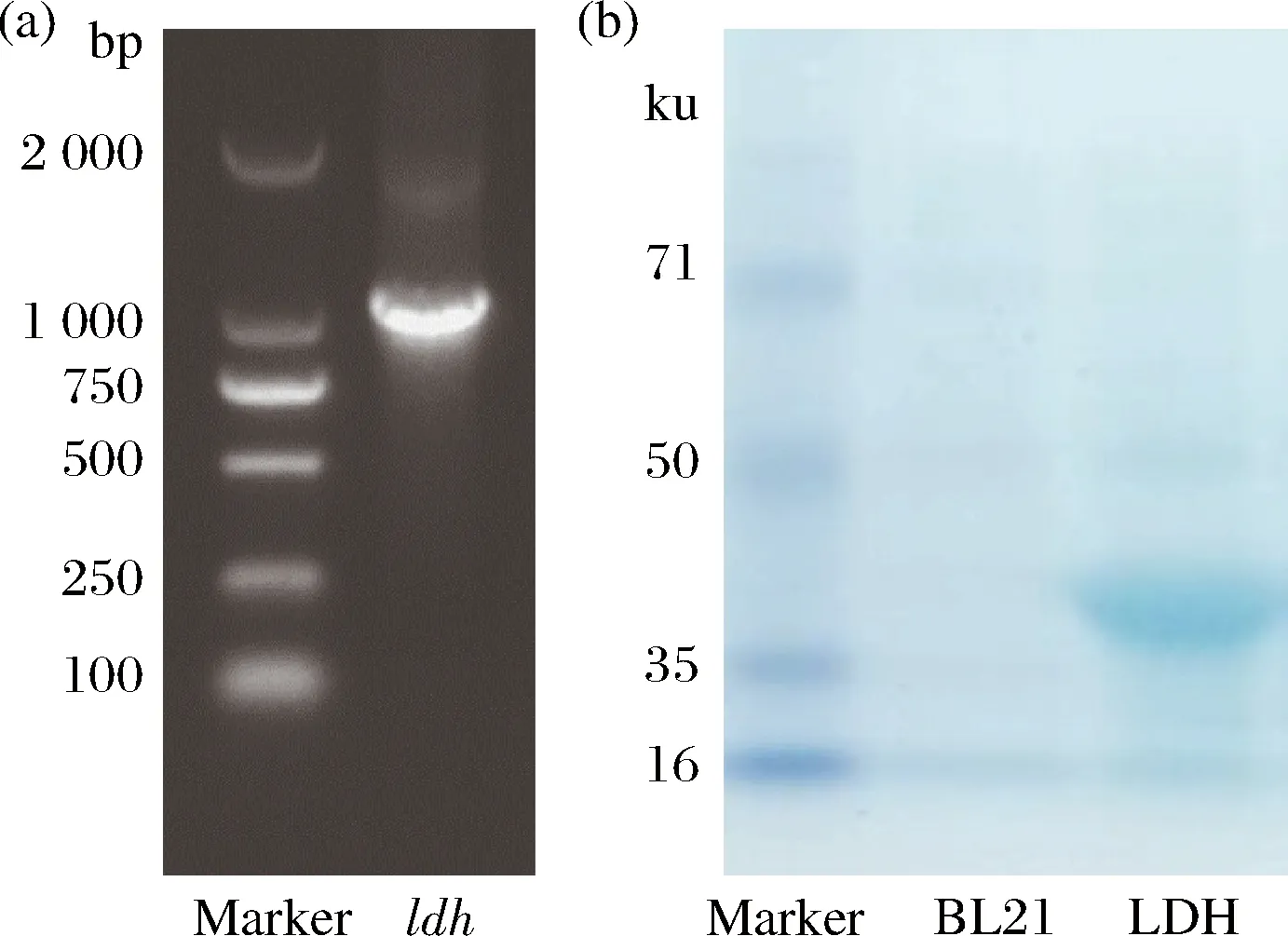
图2 PCR产物琼脂糖凝胶电泳(a);SDS-PAGE凝胶电泳(b)
Fig.2 Agarose gel electrophoresis analysis of PCR products(a);SDS-PAGE analysis of the soluble fractions of induced E. coli BL21 (DE3) and E. coli/pET28a-ldh(b)
2.2 乳酸脱氢酶重组菌表达条件优化
分别对诱导剂乳糖的用量和诱导时间做了优化实验。选择乳糖质量浓度为2~12 g/L,分别在28 ℃诱导2 h。如图3所示,随着乳糖质量浓度的升高,LDH可溶性蛋白表达量随之升高,菌体比酶活在乳糖质量浓度为8 g/L时最大,达到138.2 U/g干菌体,同时获得的生物量也较高为2.65 g/L(每1 L发酵液中含有的干菌体质量),继续升高乳糖浓度,生物量缓慢增长,但LDH可溶性表达量减少,导致重组菌比酶活下降。因此,选取8 g/L乳糖作为最佳诱导剂质量浓度。

M-Marker; 1~6-E. coli/pET28a-ldh于28 ℃分别在2、4、6、8、10、12 g/L 乳糖下诱导2 h图3 诱导剂乳糖浓度优化
Fig.3 Optimization of lactose concentration
添加8 g/L乳糖,在28 ℃下分别诱导2~10 h。如图4所示。

M-Marker;1~5-添加8 g/L乳糖分别诱导E. coli/pET28a-ldh 2、4、6、8、10 h图4 诱导时间优化
Fig.4 Optimization of induction time
随着诱导时间的延长,重组菌生物量和LDH可溶性表达量增加,重组菌比酶活提高,到8 h时达到最大值298.8 U/g干菌体,诱导10 h时,虽然生物量继续增加,但LDH可溶性表达量开始减少,重组菌比酶活随之降低。综上所述,在8 g/L乳糖质量浓度下诱导8 h为最佳诱导条件。
2.3 LDH/GDH偶联催化体系的构建与催化条件优化
乳酸脱氢酶催化的还原反应需要烟酰胺辅酶作为载体传递H+和电子,而烟酰胺辅酶价格昂贵,直接大批量添加将极大增加生产成本,因此,采用合适的方法进行辅酶再生是还原反应顺利进行的保障。将葡萄糖脱氢酶与主催化反应氧化还原酶偶联是实现辅酶高效循环的常用方法。适宜的酶组合和合适的催化条件,能够使主催化反应与辅酶再生副反应平衡,提高酶催化反应的经济性,确保还原反应的连续性。
本实验将LDH重组菌和GDH重组菌混合破碎,以葡萄糖作为氢供体构建了双酶耦联催化体系,并对催化条件进行了优化。
2.3.1 pH值对还原反应的影响
pH的变化能够引起酶分子构象的变化及酶和底物的解离状态,进而影响酶与底物的结合,使酶促反应速率发生变化。本实验控制LDH与GDH重组菌细胞质量比为1∶1,以100 mmol/L不同pH值的磷酸缓冲液悬浮并破碎,加入10 g/L PPA和葡萄糖,于30 ℃下反应,研究pH值对还原反应的影响,结果如图5-a所示。在酸性至中性范围内,产物PLA累积浓度随pH的升高而迅速增大,至pH值为7.0时最大,达到3.86 g/L。当反应液pH值处于碱性环境时,PLA累积浓度急剧下降。在所研究的pH范围内,产物ee值未发生明显变化,维持在99.9%以上。在后续实验中,维持反应液pH为7.0进行催化反应。
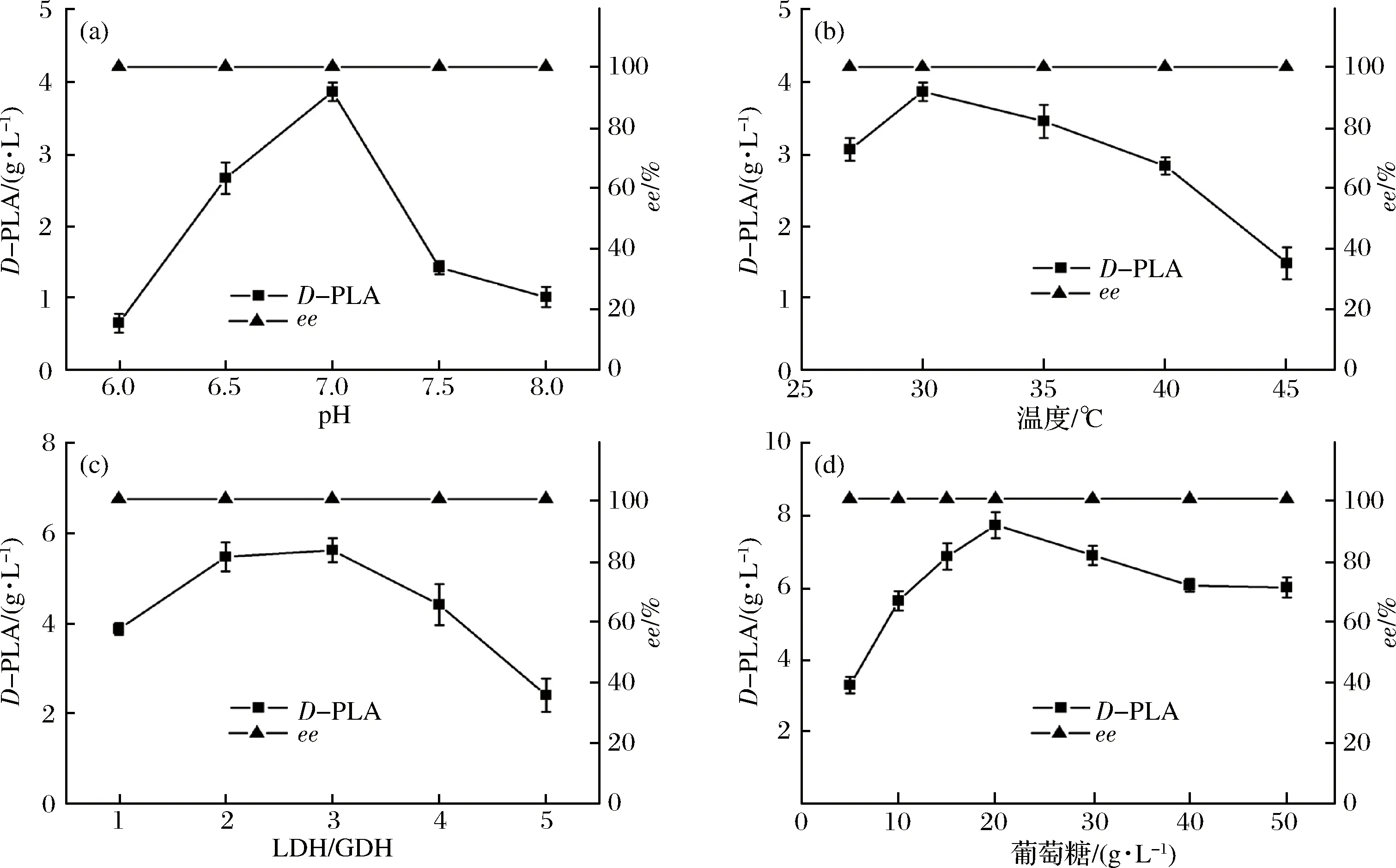
图5 催化条件优化
Fig.5 Optimization of catalytic conditions
2.3.2 温度对还原反应的影响
在一定范围内,温度的升高有助于酶分子与底物分子的有效碰撞,从而使酶促反应速率加快;而另一方面,随着温度的升高,酶分子变性加剧,活性酶分子减少,反应速率降低。酶促反应最适温度是这两方面因素平衡的结果。本实验控制LDH与GDH重组菌细胞质量比为1∶1,以100 mmol/L pH值7.0的磷酸缓冲液悬浮并破碎,加入质量浓度为10 g/L PPA和葡萄糖,在25~45 ℃范围内测定了D-PLA的产生量,结果如图5-b所示。在30 ℃时PLA累积浓度最高,达到3.86 g/L,温度升高使产物浓度降低,至45 ℃时,反应液由澄清透明逐渐变浑浊,有白色黏稠固体产生,酶变性失活加剧,产物累积质量浓度仅为1.48 g/L。在所研究的温度范围内,产物ee值未发生明显变化,维持在99.9%以上。30 ℃为双酶偶联反应体系的最适催化温度。
2.3.3 LDH与GDH重组菌细胞质量比对还原反应的影响
本实验控制反应温度为30 ℃,pH为7.0,PPA和葡萄糖浓度均为10 g/L,固定总菌体用量为20 g/L(以干菌体计),研究LDH与GDH重组菌细胞质量比对还原反应的影响,结果如图5-c所示。LDH/GDH值从1增加到3时,虽然GDH占比下降,但仍能为PPA的还原反应提供充足的还原型辅酶,LDH浓度增加使D-PLA产生速率加快,当LDH/GDH值为3时,D-PLA累积质量浓度达到最大值为5.61 g/L。继续提高质量比,GDH浓度降低,辅酶再生速率下降,LDH累积浓度降低。手性液相色谱分析可知(图6),D-PLA和L-PLA的保留时间分别约为25.9 min和28.5 min。在所研究的范围内,LDH与GDH重组菌细胞质量比对产物ee值基本无影响,始终维持在99.9%以上(图6-d)。因此,确定LDH与GDH质量比为3∶1进行后续实验。
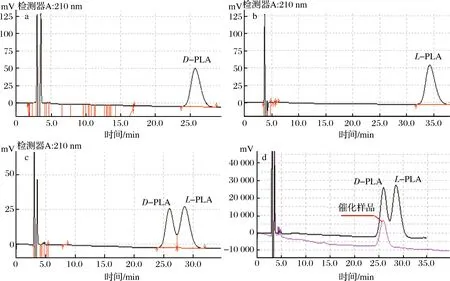
a-0.05 g/L D-PLA;b-0.05 g/L L-PLA;c-0.025 g/L D,L-PLA, d-D, L-PLA和催化样品图6 手性液相色谱图
Fig.6 Chromatogram of chiral HPLC
2.3.4 葡萄糖质量浓度对还原反应的影响
葡萄糖被GDH氧化,生成H+被烟酰胺辅酶携带,传递给PPA并使PPA还原生成D-PLA。本实验控制反应温度为30 ℃,pH为7.0,LDH与GDH重组菌细胞质量比为3∶1,PPA质量浓度为10 g/L,对葡萄糖质量浓度做了优化,结果如图5-d所示。葡萄糖质量浓度从5 g/L增加到20 g/L,D-PLA累积浓度逐步提高,至20 g/L时达到最大值7.87 g/L,在这一范围内,葡萄糖质量浓度的增加使GDH的活性提高,增加了H+的供应,从而促进了PPA的还原。继续增加葡萄糖质量浓度,D-PLA累积浓度下降,高质量浓度的葡萄糖可能抑制了GDH的活性,同时增加了反应液的黏度,增大了传质阻力。在所研究的葡萄糖浓度范围内,产物ee值未发生明显变化,维持在99.9%以上。因此,确定最佳葡萄糖质量浓度为20 g/L,在后续实验中采用2倍PPA质量浓度的葡萄糖进行催化反应。
2.4 LDH/GDH双酶耦联催化反应时间进程
在上述最优条件下,考察了不同底物浓度下双酶偶联催化反应的时间进程,结果如图7所示。
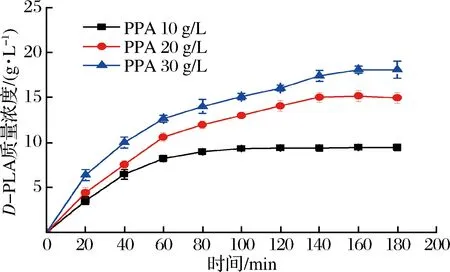
图7 不同初始底物质量浓度下的催化反应时间进程
Fig.7 Time course of asymmetric reduction
当底物质量浓度为10 g/L时,经过100 min反应基本结束,产物累积质量浓度为9.32 g/L,ee值大于99%,得率为93.2%。增加底物质量浓度,反应初速率加快,但随着产物的积累,反应速率逐渐降低,当底物质量浓度为20 g/L时,反应在140 min时基本结束,PLA累积质量浓度为15.03 g/L,ee值大于99%,得率为75.15%。当初始底物增加到30 g/L时,D-PLA最终累积质量浓度为18.09 g/L,ee值大于99%,时空产率为162.27 g/(L·d),得率降低为60.30%,表明产物质量浓度的提高抑制了还原反应的继续进行。
3 结论
本研究从Lactobacillusrossiae基因组DNA中克隆到一条编码LDH的基因,构建了E.coliBL21 (DE3)/pET28a-ldh重组菌并成功表达,通过一系列实验对LDH重组菌的表达条件进行了优化。构建了LDH/GDH双酶耦联催化体系,在实现辅酶再生的同时,促进了PPA不对称还原生成光学纯D-PLA,对催化反应条件进行了优化,D-PLA最终累积质量浓度达到18.03 g/L,时空产率为162.27 g/(L·d),显示了较好的工业化应用前景。
本实验虽取得了一定的进展,但反应过程中产物的积累对PPA还原反应产生了明显的抑制,阻碍了D-PLA得率和累积质量浓度的进一步提升。筛选合适的有机溶剂,构建双水相反应介质能有效解除产物抑制并提升底物的溶解度,有效提高反应转化率。此外,无细胞破碎液作为催化剂参与反应,使后续的产品分离纯化过程变得困难,将LDH和GDH基因导入同一宿主细胞,构建共表达基因工程菌可简化分离纯化工艺。本实验室已开展相关工作,期望进一步提升D-PLA生产强度。
