双波长高效液相色谱法同时测定肠溶片中氨基葡萄糖和吲哚美辛的含量及含量均匀度
2019-05-05张磊刘春亮王龙娟钱丹华
张磊,刘春亮,王龙娟,钱丹华
作者单位:滁州市食品药品检验中心化学室,安徽 滁州 239000
氨糖美辛肠溶片是由盐酸氨基葡萄糖和吲哚美辛两种组分按3∶1比例的复方制成的,是一种消炎镇痛药。研究显示,在非甾体抗炎药(NSAIDs)制剂中加入适当比例的氨基葡萄糖可以有效增强前者的镇痛作用[1],减少前者的用量,从而减少前者对病人造成的不适,如胃损伤[2-3]和肝损伤[4]等。氨糖美辛肠溶片现行标准有卫生部药品标准(二部)第六册,其中盐酸氨基葡萄糖含量测定采用Elson-Morgan比色法[5],操作过程易受干扰。吲哚美辛的含量测定采用提取后滴定法,专属性与重现性差。近年也有采用HPLC法测定氨糖美辛肠溶制剂中盐酸氨基葡萄糖[6-7]和吲哚美辛[8-9]含量的报道,然而能够同时高效准确测定盐酸氨基葡萄糖和吲哚美辛含量的报道很少。本研究参考美国药典最新版本(40版),采用NH2色谱柱,在双波长条件下,同时测定盐酸氨基葡萄糖和吲哚美辛的含量。本方法操作简便,耐用性良好,能够排除系统干扰,灵敏度和重复性均优于部颁标准,可用于氨糖美辛肠溶片中双组分的质量控制。
本研究起止时间为2017年9月至2018年2月。
1 仪器与试药
1.1 仪器岛津LC-20AD高效液相色谱仪(日本岛津公司),SPD-M20A PDA检测器(日本岛津公司);Hypersil NH2(5 μm,4.6 mm×250 mm)色谱柱(大连依利特分析仪器有限公司);XPE105电子天平(瑞士梅特勒-托利多集团)。
1.2 试药盐酸氨基葡萄糖对照品(中国药品生物制品检定所,批号140649-200702);吲哚美辛对照品(中国药品生物制品检定所,批号100258-200904);乙腈为色谱纯,其余试剂为分析纯(国药集团化学试剂有限公司);氨糖美辛肠溶片(A公司,批号171003;B公司,批号20161201;C公司,批号20161201)。
2 方法与结果
2.1色谱条件色谱柱:Hypersil NH2(5 μm,4.6 mm×250mm);流动相:乙腈-磷酸盐缓冲液(取3.5g磷酸氢二钾和0.25 mL氢氧化铵,加水至1000mL,用磷酸调节pH至7.5)(70∶30);流速:1.0mL/min;柱温:40 ℃;进样量为10μL,检测波长为195nm和254nm双波长测定。
2.2 对照品溶液的制备精密称取盐酸氨基葡萄糖对照品49.05 mg置25 mL量瓶中,加溶剂[乙腈-水(50∶50)]20 mL振摇使溶解并用溶剂稀释至刻度,摇匀,作为贮备液①;精密称取吲哚美辛对照品16.43 mg置于25 mL量瓶中,加溶剂20 mL振摇使溶解并用溶剂稀释至刻度,摇匀,作为贮备液②。精密量取贮备液①5 mL置于10 mL量瓶中,用溶剂稀释至刻度,作为盐酸氨基葡萄糖对照品溶液。精密量取贮备液②5 mL置于10 mL量瓶中,用溶剂稀释至刻度,作为吲哚美辛对照品溶液。精密量取贮备液①和贮备液②各5 mL混匀,作为混合对照品溶液。
2.3 供试品溶液的制备取本品20片,精密称定,研细,取约相当于盐酸氨基葡萄糖100 mg的细粉,置100 mL量瓶中,加溶剂适量,振摇使溶解并稀释至刻度,摇匀,滤过,即得。
2.4 阴性对照溶液的制备参照氨糖美辛肠溶片的处方组成,称取除盐酸氨基葡萄糖和吲哚美辛外的其它辅料成分,按供试品溶液的制备方法制备。2.5 专属性试验将以上三种溶液,在“2.1”色谱条件进样,结果显示,该条件适用于同时测定盐酸氨基葡萄糖和吲哚美辛的含量,见图1。
2.6 线性关系考察精密量取“2.2”项配置的贮备液①和贮备液②,分别用溶剂稀释,得到系列浓度为98.10、196.20、392.40、784.80、1 471.50、1 962.00 mg/L的盐酸氨基葡萄糖对照品溶液和32.86、65.72、131.44、262.88、492.90、657.20 mg/L的吲哚美辛对照品溶液,按照“2.1”色谱条件进样,在195 nm波长处记录氨基葡萄糖色谱图,在254nm波长处记录吲哚美辛色谱图,测定峰面积,以峰面积(Y)为纵坐标,浓度(X)为横坐标,分别得到线性回归方程。盐酸氨基葡萄糖标准曲线方程为:Y=601.38X+1 304.05,相关系数r=0.999 9,线性范围为98.10~1 962.00 mg/L;吲哚美辛标准曲线方程为:Y=27 995.61X-274 829.70,相关系数r=0.999 8,线性范围为32.86~657.20 mg/L。
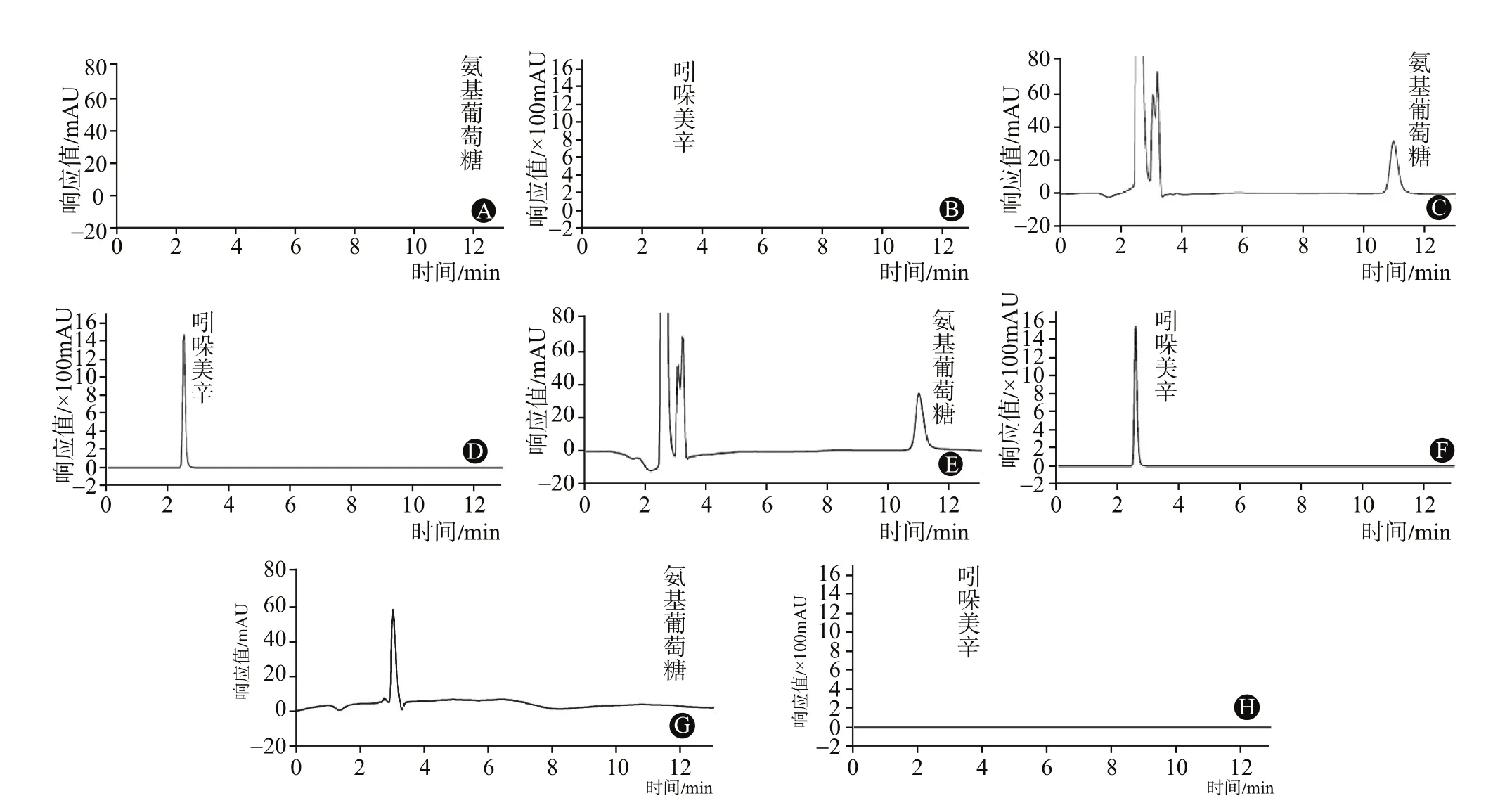
图1 双波长法测定氨糖美辛肠溶片含量的色谱图:A为盐酸氨基葡萄糖对照品(195 nm);B为吲哚美辛对照品(254 nm);C为混合对照品(195 nm);D为混合对照品(254 nm);E为样品(195 nm);F为样品(254 nm);G为阴性对照溶液(195 nm);H为阴性对照溶液(254 nm)
2.7 精密度试验取混合对照品溶液,在“2.1”色谱条件下,重复进样5次,测定峰面积,分别计算其相对标准偏差(RSD),结果氨基葡萄糖峰面积的RSD为0.64%;吲哚美辛峰面积的RSD为0.06%。表明仪器精密度良好。
2.8 重复性试验取同一批号(171003)样品,按“2.3”方法制备6份,在“2.1”色谱条件下测定,结果盐酸氨基葡萄糖含量分别为标示量的103.94%、103.88%、104.03%、102.92%、104.21%、103.92%,平均值为103.81%,RSD=0.44%(n=6);吲哚美辛含量分别为标示量的99.88%、98.68%、100.36%、100.00%、100.18%、99.86%,平均值为100.32%,RSD=0.60%(n=6),表明本方法重复性良好。
2.9 稳定性试验取同一供试品溶液,分别于0、2、4、6、8、10、12 h各进样一次,记录峰面积,结果氨基葡萄糖和吲哚美辛峰面积的RSD分别为0.59%和0.52%,表明供试品溶液在12 h内稳定。
2.10 回收率试验按供试品溶液制备方法制备样品溶液(含盐酸氨基葡萄糖2 562.4 mg/L,吲哚美辛816.1 mg/L),精密量取2 mL,共18份,分别置10 mL量瓶中,精密加入“2.2”盐酸氨基葡萄糖对照品溶液和吲哚美辛对照品溶液3、5、7 mL各3份,加溶剂稀释至刻度,摇匀,作为供试品溶液。按“2.1”色谱条件测定。计算回收率,结果见表1。平均回收率(n=9)分别为99.15%和99.28%,RSD分别为0.97%和0.66%。结果表明本方法准确度良好。
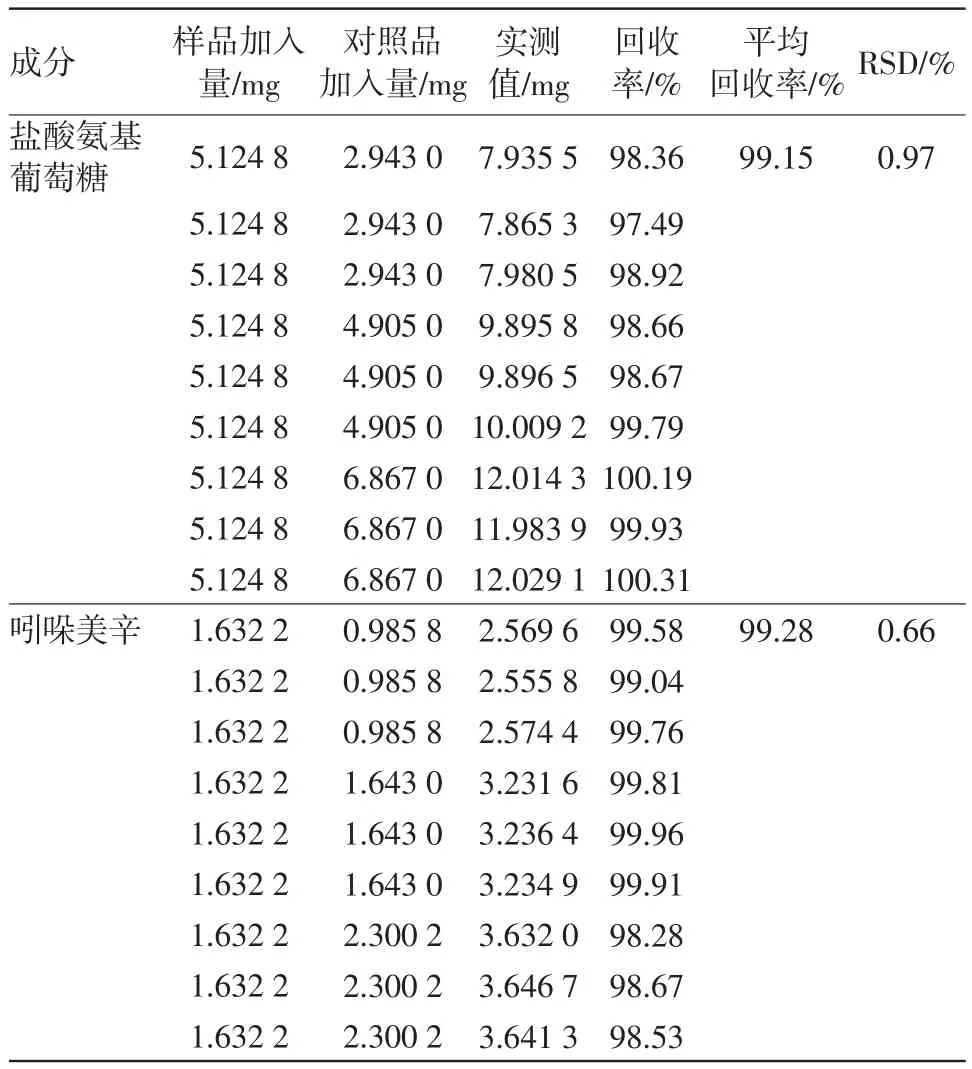
表1 加样回收率试验(n=9)
2.11 定量限取“2.2”项配置贮备液①,用溶剂稀释30倍,在“2.1”色谱条件进样,信噪比为10,溶液浓度为65.40 mg/L,确定为盐酸氨基葡萄糖对照品的定量限。取“2.2”项配置贮备液②,用溶剂稀释20 000倍,在“2.1”色谱条件进样,信噪比为10,溶液浓度为0.033 mg/L,确定为吲哚美辛对照品的定量限。
2.12 含量测定取三批样品分别依“2.3”方法制备,依“2.1”色谱条件测定,按外标法,以峰面积计算含量,并与现行部颁标准测定的盐酸氨基葡萄糖和吲哚美辛含量进行比较,结果见表2。
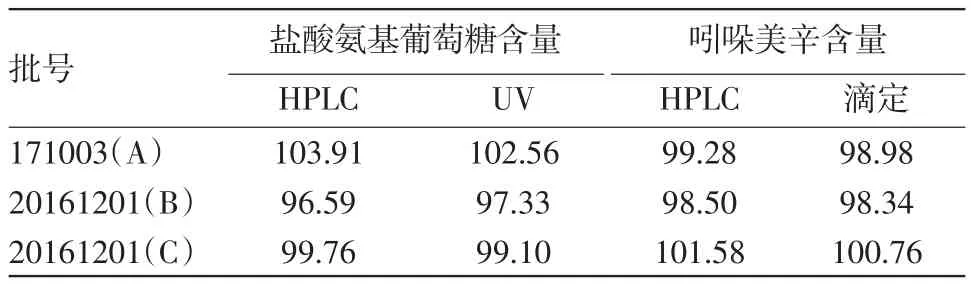
表2 氨糖美辛肠溶片含量测定结果(标示量的百分含量)/%
2.13 含量均匀度测定取样品10片,分别置100 mL量瓶中,加适量溶剂振摇使溶解,用溶剂定容至刻度。依前述方法测定每片中吲哚美辛的含量,计算A+2.2s值。结果三批吲哚美辛的A+2.2s值分别为6.0、11.8和18.5。
3 讨论
3.1 色谱柱的选择在测定盐酸氨基葡萄糖的方法中,美国药典(32版)采用C8色谱柱[10],美国药典最新版(40版)采用NH2色谱柱[11],中华人民共和国国家标准[12]采用C18色谱柱。在测定吲哚美辛的方法中,美国药典最新版(40版)采用C18色谱柱,中国药典2015年版二部采用C18色谱柱。按照上述条件进行预实验,采用C8色谱柱和C18色谱柱时,氨基葡萄糖峰的出峰时间都相对较早,其中C18色谱柱,与氯离子峰几乎完全重合,也不能与吲哚美辛峰完全分离,改变流动相的比例,氨基葡萄糖峰的保留时间几乎没有变化。考虑氨基葡萄糖是葡萄糖的衍生物,使用NH2色谱柱一般能得到较好的分离,同时参考其他糖类物质的含量测定方法[13-14],最后决定采用NH2色谱柱进行相关研究,结果表明NH2色谱柱较为适合于氨糖美辛肠溶片的含量测定。
3.2 波长的选择由于氨基葡萄糖在紫外区吸收非常弱,甚至无吸收,见图2。目前,美国药典(40版)采用195 nm波长测定。中国药典2015年版二部采用228 nm测定吲哚美辛的含量,在很多研究中,254 nm也被广泛应用于测定吲哚美辛的含量[15-16]。按照上述条件进行预实验,采用195 nm能较好地实现氨基葡萄糖峰的分离。在195 nm条件下,吲哚美辛峰易受氯离子峰干扰,故不宜采用195 nm同时测定双组分的含量。在228 nm条件下,吲哚美辛峰也易被氯离子峰轻微干扰。结合吲哚美辛峰光谱图,见图3,本实验选用254 nm。在此条件下,氯离子几乎无吸收,不会对吲哚美辛峰造成干扰。因此,本研究最终采用195 nm作为氨基葡萄糖检测波长,254 nm作为吲哚美辛检测波长。
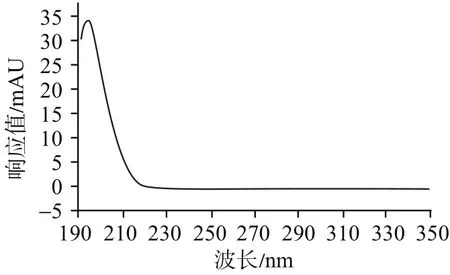
图2 氨基葡萄糖峰光谱图
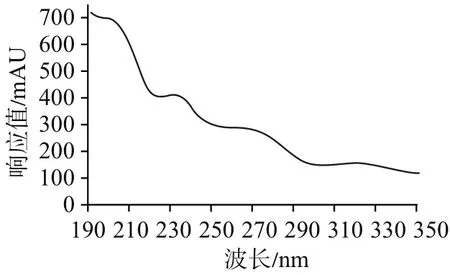
图3 吲哚美辛峰光谱图
3.3 流动相的选择本研究的重点考虑因素是氯离子峰和氨基葡萄糖峰的有效分离。美国药典(32版)以磷酸盐缓冲液(0.05%磷酸,用氢氧化钾调节pH至3.0)-乙腈(40∶60)为流动相。在实际试验中,由于该流动相水相比例较高,影响NH2色谱柱的使用寿命,重复进样后,理论板数迅速下降,拖尾因子迅速上升。因本研究采用NH2色谱柱,为延长色谱柱寿命,提高耐用性,在流动相中适当提高乙腈比例,在亲水作用色谱模式下,使用较高比例的乙腈来保留亲水化合物,并加入磷酸氢二钾以改善峰型,通过氢氧化铵调节pH值使氨基葡萄糖呈弱阴离子化合物,最终确定流动相为乙腈-磷酸盐缓冲液(取3.5 g磷酸氢二钾和0.25 mL氢氧化铵,加水至1 000 mL,用磷酸调节pH至7.5)(70∶30)。采用该流动相,在195 nm条件下,氨基葡萄糖保留时间约为11.0 min,氯离子保留时间约为3.2 min,实现了氯离子与氨基葡萄糖的有效分离。在254 nm条件下,吲哚美辛保留时间约为2.6 min。该系统下,氨基葡萄糖与吲哚美辛理论板数保持稳定,峰形良好。结果显示,本次试验选定的氨糖美辛肠溶片含量测定方法耐用性良好。
3.4 含量均匀度的控制中国药典2015年版四部规定,单剂标示量小于25 mg或主药含量小于每个单剂体质量25%的片剂应检查含量均匀度。氨糖美辛肠溶片每片含吲哚美辛25 mg,且小于每个单剂体质量的25%,应控制其含量均匀度。本研究考察了3个企业各1批氨糖美辛肠溶片的吲哚美辛含量均匀度。其中1个企业的一批样品含量均匀度A+2.2s值大于15.0。由于现行标准中未规定氨糖美辛肠溶片的含量均匀度要求,生产企业在混料的过程中的可能会存在一定问题。因此在标准中增加含量均匀度检查项,对于产品质量控制,全面反映产品的质量优劣是有必要的。3.5 小结本实验采用双波长HPLC检测法,建立了同时测定氨糖美辛肠溶片中盐酸氨基葡萄糖和吲哚美辛含量及含量均匀度的方法。与现行国家标准相比,具有专属性强,操作简便快速等优势,且本方法稳定性高、线性范围宽、结果准确可靠,可作为氨糖美辛肠溶片质量控制的方法,为其质量标准的修订提供了有价值的参考。
