光动力新药帕利泊芬研究进展
2019-04-24闵祥燕曹宁严懿嘉陈志龙
闵祥燕,曹宁,严懿嘉,陈志龙*
(1. 东华大学化学与生物学院,上海 201620;2. 上海先辉医药科技有限公司,上海 200433)
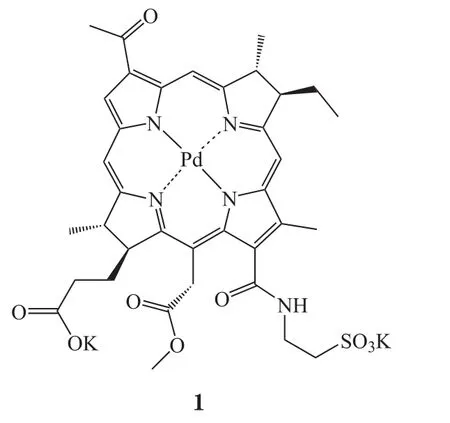
光动力疗法(photodynamic therapy,PDT) 是一种微创、治疗浅表性肿瘤的新方法,是将光敏剂(photosensitizer,PS)定位于靶组织上,PS吸收特定波长的光后,发生光化学反应,进而杀伤和消灭目标位置上的肿瘤细胞。帕利泊芬(1,padelipor fin,WST11,Tookad®Soluble,Stakel®)是一种与钯配位的细菌叶绿素衍生物,分子式:C37H41N5O9PdS(CAS:698393-30-5),由 Memorial Sloan-Kettering Cancer Center、Steba Biotech 和 Weizmann Institute of Science 这 3 家公司联合研制,已于2017年11月获欧洲药品管理局(EMA)批准上市,作为进行血管靶向PDT(vasculartargeted PDT,VTP)的PS,用于治疗前列腺癌;其用于治疗肾癌的研究处于Ⅱ期临床阶段。本品具有水溶性,静脉给药后,可靶向聚集于肿瘤组织,被适当波长的光激活并产生大量活性氧(ROS),进而诱导肿瘤细胞发生凋亡或坏死。本文主要对帕利泊芬的作用机制、合成路线、药理作用以及临床研究进行介绍。
1 帕利泊芬作用机制
PS是一类在光照射下能产生ROS的化合物,理想的PS应具有水溶性。此外,其激发波长应在PDT光学“窗口”(650~850nm)内,能最大化地穿透组织[1-3]。光动力疗法主要有2种作用机制:激发态PS(S1)通过系间窜越将能量转移到激发三重态(T1),并通过电子或质子转移过程直接与基质或溶剂反应,产生自由基和超氧阴离子,超氧阴离子可进一步反应生成过氧化氢(Ⅰ型反应);此外激发三重态PS也可以产生单线态氧(1O2),1O2可以通过诱导细胞凋亡或坏死直接杀死肿瘤细胞(Ⅱ型反应)[4-6]。若超氧阴离子浓度过高,过氧化氢会与超氧阴离子反应,生成具有更高活性的羟基自由基,而羟基自由基在细胞内可以进攻和氧化任意的生物分子。此外也可能是由超氧阴离子与周围的铁离子或铜离子之间发生芬顿反应(Fenton’s reaction),产生羟基自由基[6-10]。帕利泊芬作为一种高效的PS,在水溶液中经历Ⅰ型反应机制[11],产生了带电或中性自由基,这些自由基迅速与氧分子反应生成ROS,从而杀死肿瘤细胞。光敏剂作用机制如图1所示。

图1 光敏剂作用机制Figure 1 Mechanism of photosensitizer
2 帕利泊芬的发现历程及其合成路线
在帕利泊芬问世之前,已有数个卟啉类药物被用于肿瘤的临床治疗。血卟啉衍生物Photo fin是全球首个获批准上市的光敏剂,随后与Photo fin结构相近的卟啉衍生物喜泊芬及Photogem等也先后在俄罗斯、德国和中国上市。然而,这类光敏剂存在一些缺点:首先,其吸收波长为410nm,接近太阳光的辐射波长(475nm),且光敏剂在体内滞留时间较长,PDT治疗后患者会出现明显皮肤光毒性,病人需在治疗后避光28~42d;其次,治疗使用的可见光的组织穿透强度较弱,治疗深度浅;另外,此类PS摩尔吸光系数小,因此往往需要提高药物剂量或光剂量来达到治疗效果,毒副作用大。
近年来,研究人员将注意力转向了叶绿素和细菌叶绿素的衍生物,其具有特定的吸收光谱区域(650~850nm)和激活后能快速降解的特性,被认为是两类更具优势的PS。与叶绿素类衍生物相比,细菌叶绿素衍生物是一类具有2个不饱和吡咯环的四吡咯大环,其通常在近红外(NIR)范围内的750nm处的PDT光学窗口内具有Qy吸收带,在近红外光区组织渗透至8mm[12]。不过,细菌叶绿素衍生物在常温常压下不稳定,且与血卟啉衍生物相比,其单线态氧的产量较低(由中心金属离子决定)[13]。为了增加细菌叶绿素衍生物在常温常压下的稳定性,Avigdor等[13]尝试将其中心原子Mg替换为2价的Pd、Pt、Sn、Ni、Cu、Zn和Mn等。该课题组发现,中心原子Mg被Pd取代后,可显著增加细菌叶绿素大环的氧化电位,同时可以大大提高分子的跨系统速率,使其达到激发三重态。Pd-细菌叶绿素衍生物的生物分布和药动学研究显示,其仅保留在血液循环中且在光照后的保留时间很短,解决了病人在PDT后需长期避光的问题。然而,Pd-细菌叶绿素衍生物在水溶液中溶解度低,配制注射药物时,需要使用增溶剂(如氢化蓖麻油)来增加其溶解性,使用大剂量的增溶剂曾在小鼠实验中引发了诸多不良反应(如呼吸急促、萎靡不振等)。因此,增加Pd-细菌叶绿素衍生物的水溶性是迫切需要解决的另一个问题。
此前,有研究发现细菌叶绿素a(2)的17位碳原子位置被氨基酸、多肽、蛋白质等多种分子取代后,可增加化合物的亲水性[14]。Rosenbach-Belkin等[15]研究发现,细菌叶绿素的丝氨酸衍生物水溶性显著改善,且在小鼠实验中显示出较快的血液循环清除速率。遗憾的是,丝氨酸-细菌叶绿素衍生物极不稳定,能被光迅速氧化。但这也为Avigdor等研究如何提高Pd-细菌叶绿素衍生物溶解度提供了有意义的参考,即在Pd-细菌叶绿素衍生物17位碳原子位置引入亲水基团,如COO-、COS-、SO3-和 PO32-等[12]。在 Pd-细菌叶绿素衍生物的17位碳原子位置引入SO3-,即制得帕利泊芬,其可直接溶于水。此外,生物分布和药动学分析研究亦获得了令人满意的结果。在荷有M2R黑素瘤和HT-29结肠癌的模型小鼠中进行的研究证实帕利泊芬具有显著抗肿瘤作用,另外,该药物在动物体内的清除速率快,是非常理想的血管靶向PS。
图2介绍了帕利泊芬及其前体药物padopor fin(3,WST09)的合成路线。值得一提的是,padopor fin也曾进入前列腺癌治疗的临床阶段,但研究结果显示其会引发低血压等副作用,且副作用较大[16-21]。根据文献方法,首先从球形细菌的培养物中提取得到原料细菌叶绿素a,通过稀盐酸脱去配位金属Mg,得到化合物4。随后将醋酸钯加入到化合物4的二氯甲烷/甲醇混合溶液中,在氩气保护下室温过夜搅拌,鳌合形成padopor fin[22]。最后,将padopor fin完全溶解于DMSO中,并加入催化当量的1,3-二环己基碳二亚胺(DCC)和4-二甲氨基吡啶(DMAP),最后添加适量溶解牛磺酸的K2SO3溶液(pH=8.2),在氮气保护条件下室温避光搅拌4d,最终得到13-C位置开环的产物,即帕利泊芬[13,23]。
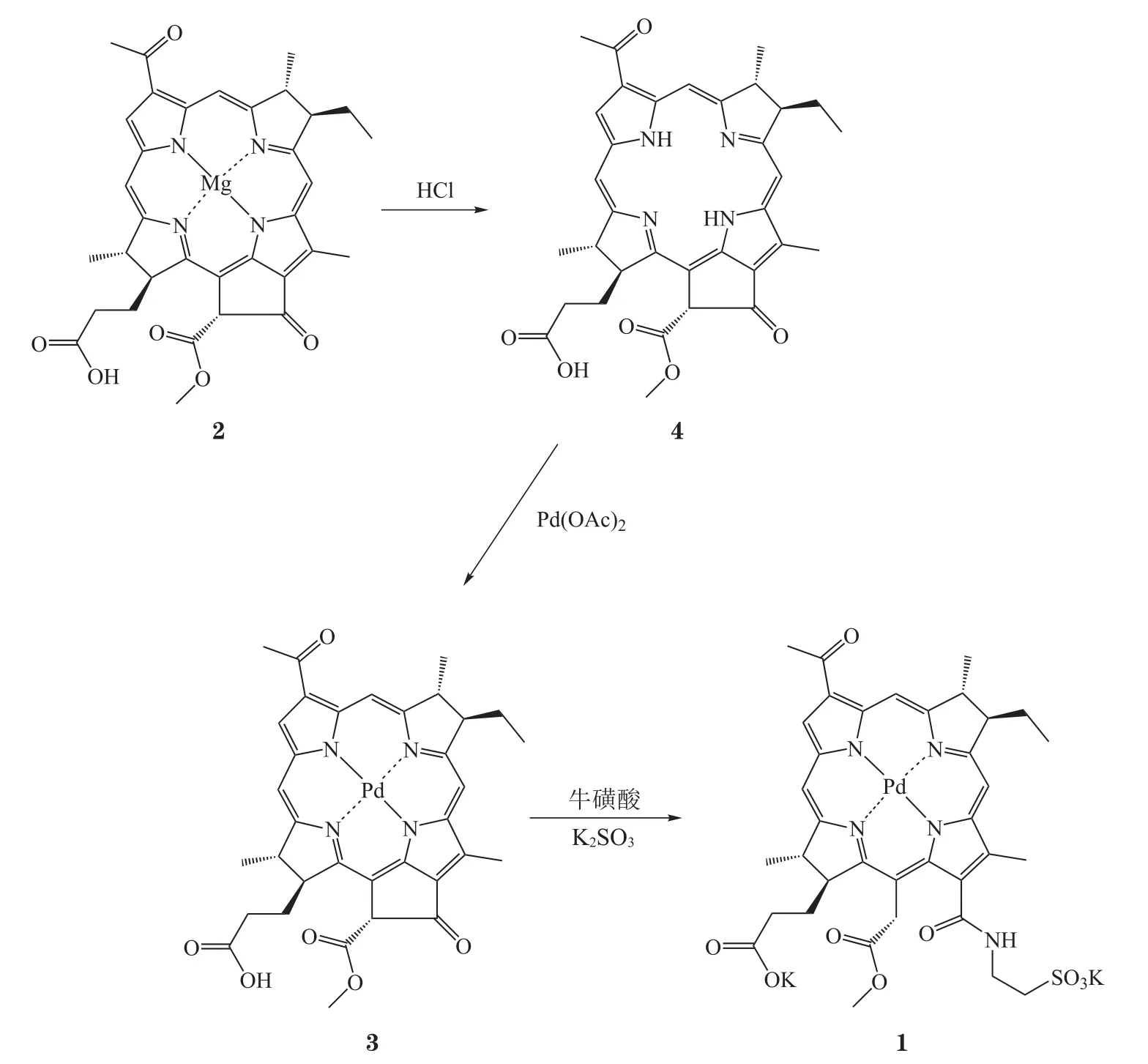
图2 帕利泊芬的合成线路Figure 2 Synthetic route of padeliporfin
3 帕利泊芬的药理学研究
3.1 体外研究
3.1.1 细胞对帕利泊芬的摄取与细菌叶绿素衍生物的性质类似,帕利泊芬呈现出非常弱的荧光,因此不能通过荧光法来对细胞摄取帕利泊芬进行定量检测[24]。每个帕利泊芬分子结构中都含有一个Pd原子,且生理条件下吡咯环结构螯合的Pd离子非常稳定,因此,可以通过测定细胞中的Pd原子含量,来表征细胞对帕利泊芬的摄取量[24]。
Posen等[25]将小鼠黑素瘤单层细胞分别与浓度为0、15、30、60 和 100nmol·L-1的帕利泊芬溶液一起孵育 4h,然后用功率为20mW的激光器照射10min。光照后立即用电感耦合等离子体质谱法(ICP-MS)测定细胞内Pd原子的含量,从而确定细胞内帕利泊芬的含量。结果显示,随着帕利泊芬浓度的递增,检测到细胞内Pd原子的含量也递增。光照后细胞继续培养24h,然后进行噻唑蓝(MTT)检测,测定细胞的存活率。结果表明,随着帕利泊芬浓度的递增,细胞摄取到的帕利泊芬也增多,胞内帕利泊芬在光照下被激活后,细胞存活率随溶液中帕利泊芬浓度递增而降低(见图3)。
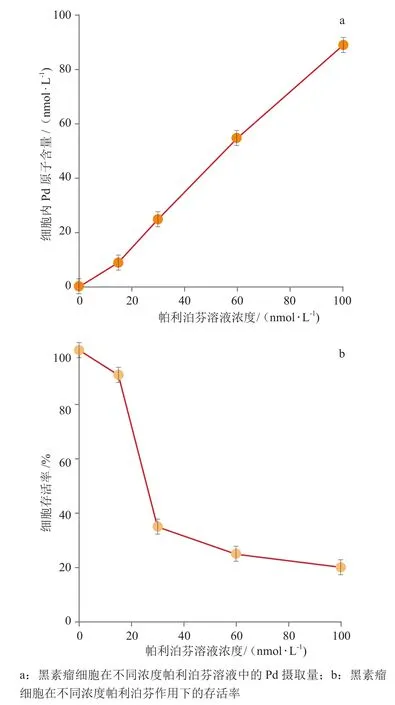
图3 黑素瘤细胞在不同浓度帕利泊芬溶液中的Pd摄取量及光照后存活率Figure 3 The Pd uptake and survival rate of melanoma cells at different concentrations of padeliporfin
Brandis等[23]研究了温度和血清等因素对细胞摄取帕利泊芬的影响,发现在37℃时,细胞中帕利泊芬浓度在最初 5~10min 内迅速增加,于培养开始后的 50~60min趋于稳定;而温度降至4℃时,帕利泊芬能够迅速渗透入细胞并在培养10min后胞内浓度达到平稳;在没有血清的情况下帕利泊芬能形成直径为1~2nm的小聚集体,但小聚集体并不影响细胞膜的吸附[26]。黑色素瘤细胞摄取帕利泊芬的实验结果表明,帕利泊芬在细胞中的摄取是由血清白蛋白运输介导的。Mazor等[24]也证明了帕利泊芬与血清白蛋白络合会促进细胞摄取帕利泊芬。
3.1.2 帕利泊芬的细胞毒性研究在细胞PDT实验中,随着帕利泊芬浓度的增加,细胞存活率递减。为了确定是单纯的高浓度药物杀死了细胞,还是药物发生光化学反应后杀死细胞,Posen等[25]将小鼠胚胎心脏内皮细胞(H5V)分别与不同浓度(0、10、100和1000nmol·L-1)的帕利泊芬一起孵育 4h 后,分别用功率为20mW的激光器照射10min以及避光处理,然后细胞继续培养24h,分别对细胞进行MTT检测,测定细胞的存活率。研究显示,光照处理的细胞,随着培养物中帕利泊芬浓度的增加,细胞存活率下降,半数致死剂量(LD50)约为 30nmol·L-1;相反,对于避光处理的细胞,帕利泊芬浓度对细胞存活率影响不明显(见图4)。因此细胞摄取到的帕利泊芬,只有在被光激活后才会杀死细胞且细胞存活率随帕利泊芬浓度递增而降低。即帕利泊芬对细胞的光毒性明显,而暗毒性很小。

图4 帕利泊芬在光照和避光条件下的细胞毒性Figure 4 Cytotoxicity of padeliporfin under light and dark conditions
3.2 动物体内研究
利用肿瘤动物模型进行PDT实验是筛选抗肿瘤PS的常用手段之一。Mazor等[24]将黑素瘤模型小鼠分为实验组和对照组,给实验组小鼠尾静脉注射帕利泊芬(6mg·kg-1),然后用 45J·cm-2的激光照射,经皮照射肿瘤部位5min;对照组小鼠接受给药不照光、照光不给药或不给药亦不照光的处理方式。结果显示,接受不同处理的对照组小鼠,其肿瘤生长速率相似,且尾静脉注射30min后未观察到小鼠出现皮肤光毒性。在PDT实验后,所有实验组小鼠经治疗的肿瘤部位在24~48h内均发生肿瘤组织的坏死,小鼠继续养殖14d后原肿瘤部位均变平,到90d后约有60%的小鼠被治愈。
此外Fleshker等[27]也开展了类似研究,对荷有CT26 结肠癌的小鼠尾静脉注射帕利泊芬(6mg·kg-1),然后进行PDT治疗。治疗后,向小鼠体内注射可识别肿瘤的荧光素,再用生物荧光成像(bioluminescence imaging,BLI)监测每只小鼠的肿瘤变化情况,若BLI信号为阴性则说明小鼠体内没有肿瘤,若BLI信号为阳性则说明小鼠体内有肿瘤。实验中共有53只小鼠接受PDT,其中有40例BLI信号为阴性,有39例在随后的90d观察期间未观察到BLI阳性信号,仅有1例在PDT后第30d死亡(未见肿瘤再生),即PDT后约75%(40/53)的小鼠肿瘤治愈,其余13只小鼠PDT24h后,观察到BLI阳性信号,且在随后的观察期间可见局部肿瘤复发。实验结果表明,帕利泊芬治疗结肠癌CT26荷瘤小鼠的疗效显著。
4 帕利泊芬治疗前列腺癌的临床应用
帕利泊芬作为治疗前列腺癌的新型血管靶向PS,已成为PDT研究领域近年来的热点。临床上,全身麻醉的前列腺癌患者接受静脉注射帕利泊芬(4mg·kg-1),同时,经直肠和会阴将装有超声光缆的中空塑料针置于患者前列腺内,用光纤功率为150mW的激光照射,其输送的纤维数量和总光能随肿瘤位置和前列腺体积而变化[28],可用于局部前列腺癌的治疗。
Lebdai等[26]对82名前列腺癌患者静脉注射帕利泊芬(4mg·kg-1)后进行 PDT 治疗,治疗 6 个月后对所有患者进行前列腺活检。结果显示,有62名(76%)患者活检结果为阴性,20名患者活检结果为阳性,其中,有9名(11%)患者为临床显著癌(clinically signi ficant cancer),另外的11名(13%)患者为临床非显著癌(non-clinically signi ficant cancer)。此外,Azzouzi等[29]也开展了类似的临床研究,对114名前列腺癌患者进行PDT,治疗6个月后对所有患者进行前列腺活检,其中有78名(68.4%)患者的活检结果为阴性。上述临床研究结果均证实帕利泊芬治疗前列腺癌疗效显著。
5 帕利泊芬治疗前列腺癌的优势
5.1 水溶性好,靶向性高
帕利泊芬是一种水溶性的血管靶向PS,通过血液循环可直接靶向聚集于前列腺癌细胞,并在癌细胞内被激活,从而产生大量的ROS,发挥其治疗前列腺癌的功能[30]。
5.2 代谢快,体内无蓄积
Brandis等[23]研究了帕利泊芬在小鼠和大鼠中的药动学特性,发现帕利泊芬仅保留在血液循环中,且血液中的保留时间非常短。Mazor等[24]对小鼠尾静脉注射帕利泊芬(6mg·kg-1),并在此后的不同时间点,分别测定小鼠心、肝、脾、肺、肾以及血液中的药物浓度。结果显示,尾静脉注射2min后,血液中帕利泊芬的含量最高,在注射后5min仅在肝、脾、肺、肾和血液中发现存有帕利泊芬。给药24h后,所有组织中均检测不到帕利泊芬。帕利泊芬能在组织中快速清除可能与引入的牛磺酸基团有关[31]。牛磺酸是细胞液中的β-氨基酸,广泛分布于哺乳动物的肝脏中,参与肝脏的解毒。在解毒过程中,牛磺酸与肝脏中不同分子结合可增加药物的水溶性,从而促进了肝脏对药物的清除[32]。图7显示了帕利泊芬在小鼠体内的生物分布情况。研究人员发现帕利泊芬可以从血液中快速消除,不会在皮肤组织中积聚,从而确保帕利泊芬具有较低的皮肤光毒性[33]。
5.3 安全性高
Lebdai等[26]使用IPSS和IIEF-5问卷分别评估了前列腺癌患者在PDT治疗后的泌尿系统症状和勃起功能。40名前列腺癌患者经PDT治疗后,其中有38名患者在治疗后第1d无需导尿管,另外2名患者在PDT治疗几天后也无需导尿管。临床研究结果证明帕利泊芬不会引起患者的心血管毒性或肝酶改变、术中麻醉问题、直肠瘘或皮肤光敏等,IPSS评分和IIEF-5评分也未受较大影响。综合评价认为,帕利泊芬在临床使用方面具有良好的安全性。
6 结语与展望
随着新型PS的不断开发,PDT在临床中的应用越来越广泛。帕利泊芬作为一种水溶性的血管靶向PS,可直接靶向聚集于肿瘤细胞,在肿瘤细胞内被激活,发生一系列的光化学反应从而发挥抗肿瘤疗效。目前研究认为,帕利泊芬治疗前列腺癌有较好疗效,但患者经PDT后会产生血尿、会阴疼痛和射精失败等副作用,还需要其他辅助疗法来消除这些副作用。因此帕利泊芬治疗前列腺癌引起的不良反应还需要进一步的深入研究,这将有助于帕利泊芬的合理应用,对其能够安全有效地用于治疗前列腺癌具有重要的指导意义。随着对帕利泊芬治疗前列腺癌的药理作用和机制的深入研究以及在临床上的应用,人们对PDT的认知将更为全面,可以相信,未来将会有更多的PS被研发出来,在肿瘤的临床治疗中发挥重要作用。

图5 帕利泊芬给药后在小鼠体内的生物分布Figure 5 Biodistribution of padeliporfin in mice after administration
