蛋白多肽类药物长效化技术研究进展
2019-04-24刘梦于彭城徐寒梅
刘梦,于彭城,徐寒梅
(中国药科大学江苏省合成多肽药物发现与评价工程研究中心,江苏 南京211198)
蛋白多肽类药物毒性小、特异性强,针对某些疾病的药理活性较好,在药物开发中被认为是十分有前途的生物大分子,但此类药物稳定性较低、易被体内各种酶降解以及肾清除迅速,导致其半衰期较短,从而限制了药物的使用。因此,开展蛋白多肽类药物长效化相关研究十分重要,延长其血浆半衰期的策略不仅可以改善既定药物的药动学特性,也可能为药物使用开发新的适应证。本文基于蛋白多肽类药物药动学特点,综述了蛋白多肽类药物长效化技术的研究进展。
1 蛋白多肽类药物药动学特点
1.1 吸收
蛋白多肽类药物因相对分子质量的不同,其吸收方式也存在一定差异。通常,小肽的吸收方式包括载体转运和被动扩散;大分子蛋白多肽的吸收方式包括膜脂扩散以及细胞间隙扩散;高度亲脂性药物则能通过淋巴途径吸收进入体内[1]。由于胃肠道蛋白降解酶对于蛋白多肽类药物降解作用的影响,此类药物口服给药生物利用度较低,多数都优先选择注射给药,但也因此影响了患者的用药顺应性。研究者一直致力于开发适合蛋白多肽药物的非注射给药途径,目前有望通过纳米技术克服这一瓶颈[2]。除了联合给药及对药物进行改造,目前还开发了多种技术模型来研究及预测蛋白多肽类药物在体内的肠道吸收及其体内生物利用度[3]。皮下注射是蛋白多肽药物重要的给药途径,此种给药方式吸收过程较为缓慢,能维持药物在体内稳定的效应,并且可以避开肝首过效应,降低药物的给药剂量[4]。
1.2 分布
蛋白多肽类药物在体内的分布主要由扩散和较小程度的对流外渗组合驱动,分布体积通常不大于细胞外体液的体积[5]。另外,此类药物体内分布具有明显的靶向性,根据此特点可开发靶向给药,使药物能够快速到达病灶部位发挥药物疗效。Han等[6]利用丙氨酰-谷氨酰-酪氨酰-亮氨酰-精氨酸序列(AEYLR)作为体外靶向表皮生长因子受体(EGFR)的小肽配体,研究了AEYLR和AEYLR共轭纳米结构脂质载体(NLC)的体内靶向性。结果表明,AEYLR修饰的NLC是有希望用于联合癌症化学疗法的靶向递送系统,不仅能提高治疗效果,也能减少毒副作用。研究人员对切割人血清白蛋白得到的白蛋白片段进行光学成像研究,发现其可选择性地积聚在肾脏中,可作为肾靶向的预期载体[7]。精氨酰-甘氨酰-天冬氨酸序列(RGD)与包封有荧光染料吲哚菁绿(ICG)的聚乙二醇(PEG)化脂质体的缀合物,可作为一种有效的递送系统靶向胃癌细胞中高表达的整合素受体[8]。
1.3 代谢和消除
血液、肝脏、肾脏和小肠中含有各种蛋白酶和肽酶,是蛋白多肽类药物发生降解的主要部位。除此之外,因蛋白酶和肽酶也存在于全身其他部位,此类药物的代谢并不限于经典的消除器官,大多数半衰期短的多肽主要经过肽酶快速水解被消除。通常,相对分子质量较小的多肽可以通过肾脏中的肾小球自由过滤,然后在近端小管细胞的刷状缘膜中水解降解。理论研究和临床数据表明,对于蛋白类治疗剂,肾脏可在一定程度上调节这些物质的功效或安全性,并在分解代谢中发挥相关作用[9]。若机体对蛋白水解具有抗性,则主要通过肾脏消除。例如,艾塞那肽是胰高血糖素样肽-1(GLP-1)模拟物,含有39个氨基酸,氨基酸取代使艾塞那肽对二肽基肽酶-4的降解产生抗性,因此艾塞那肽经肾小球滤过后在肾小管中酶促降解[10]。
肝脏代谢可能主要对一些小肽药物的代谢消除具有实质性作用,例如用于治疗多发性骨髓瘤的二肽硼替佐米会经历广泛的氧化性肝代谢,但若机体肝功能发生损伤,则清除率会降低[11]。类似地,环状十一肽环孢素通过被动扩散进入肝细胞,几乎完全经过肝脏代谢消除[12]。
蛋白多肽类药物还具有非线性药动学特征:大部分药物与药理学靶标高度结合,随后消除药物-靶标复合物,由于受体的数量有限,会出现剂量依赖性非线性药动学,如血小板生成素模拟肽靶向介导药物的处置过程显示出明显的剂量依赖性[13]。
2 改善蛋白多肽类药物半衰期的方式
天然的蛋白多肽类药物通常具有较差的吸收、分布、代谢和排泄特性,如半衰期短、渗透性低,有时溶解度也较低[14]。为了更加充分地发挥药物的治疗效果,提高患者的耐受性,目前已经开发出多种改善蛋白多肽类药物稳定性的策略,延长体内半衰期,使其长效化。
2.1 氨基酸替代
蛋白多肽类药物含有的某些代谢不稳定的氨基酸,会极大地影响药物的半衰期,因此取代这类代谢不稳定的氨基酸是延长药物半衰期的一种方式。生长抑素是一种多肽类药物,含有14个氨基酸,其体内半衰期非常短,只有2~3min,研究发现,通过将第8位的D-色氨酸用L-色氨酸取代可以增加作用时间,延长其体内半衰期[15]。
除一般氨基酸取代策略,将非天然氨基酸引进蛋白多肽类药物中也可以增加此类药物的稳定性、延长半衰期,目前已研究出多种将非天然氨基酸引入蛋白多肽中的方法[16-17]。促性腺素释放激素(GnRH)本身的血浆半衰期非常短,而合成的十肽GnRH拮抗剂(西曲瑞克、地加瑞克、阿巴瑞克)和GnRH激动剂(曲普瑞林、亮丙瑞林、布舍瑞林、戈舍瑞林、那法瑞林)均含有非天然氨基酸,与GnRH相比均表现出延长的半衰期[18]。同时,引入非天然氨基酸还可提高肽的亲和力[19]。目前,已有多种治疗性多肽通过氨基酸替代有效延长了体内半衰期(见表1)。
非天然氨基酸取代策略的一个主要问题是非天然氨基酸的潜在毒性。例如,具有非天然氨基酸取代的GnRH类似物与患者的药物不良反应相关[20]。另有研究发现,非天然氨基酸会在肝脏及其他器官中积聚[21]。
2.2 定点修饰突变
突变蛋白分子的特定位点,也能够改善其稳定性并延长蛋白多肽类药物的血浆半衰期。如人胰岛素,将A链第21位的天冬氨酸突变为甘氨酸,在B链C端第30位加入2个精氨酸,得到的突变体在体内能持续释放且血药浓度保持恒定,长达24h[22]。抗制瘤素M(OSM)是一种相对分子质量为28000的可溶性细胞因子,由活化的免疫细胞分泌,越来越多的证据表明,OSM在炎症中起着关键作用。CNTO8212是抗OSM IgG1单克隆抗体Fc区突变体,药动学研究结果显示,CNTO8212具有较低的全身清除率和较长的食蟹猴体内终末半衰期,预测的人药动学参数提示CNTO8212可在临床中以较低频率给药[23]。
2.3 通过基因手段与其他蛋白融合
融合蛋白与蛋白多肽类药物基因融合表达,能够增加药物的相对分子质量,降低体内肾清除率,可达到延长药物体内半衰期的目的。FcIII是一种13-mer IgGFc结构域结合肽(IgGBP),通过融合蛋白结合血清IgG来增加蛋白质的体内半衰期,其与未融合修饰的蛋白质相比,半衰期增加了75倍[24]。IgG1Fc片段非糖基化单链与干扰素-α(IFN-α)融合后得到IFN-α/Fc-MD,与商业聚乙二醇化IFN-α(PEG-IFN-α)进行比较,IFN-α/Fc-MD显示出与PEG-IFN-α相当的抗病毒活性。体内药动学测定显示,IFN-α/Fc-MD在SD大鼠中具有比PEG-IFN-α更长的半衰期[25]。水蛭素与白蛋白融合后,药物体内作用时间延长,且其生物学活性也得到增强[26]。艾塞那肽被广泛用于治疗2型糖尿病,将艾塞那肽与人血清白蛋白(HSA)结合得到的融合肽显示出与天然艾塞那肽类似的促胰岛素分泌活性。皮下给药后艾塞那肽-HSA的药动学分析显示,其与艾塞那肽相比,血浆半衰期延长了4倍。此外,艾塞那肽-HSA在口服葡糖糖耐量试验中显示出显著改善的抗高血糖作用且增强了降血糖作用[27]。通过基因融合技术提高蛋白多肽类药物稳定性,是目前蛋白多肽类药物长效化的良好途径,其中不少融合蛋白药物已经被批准上市(见表2),同时也有相当多的融合蛋白药物处于临床在研阶段(见表3)。
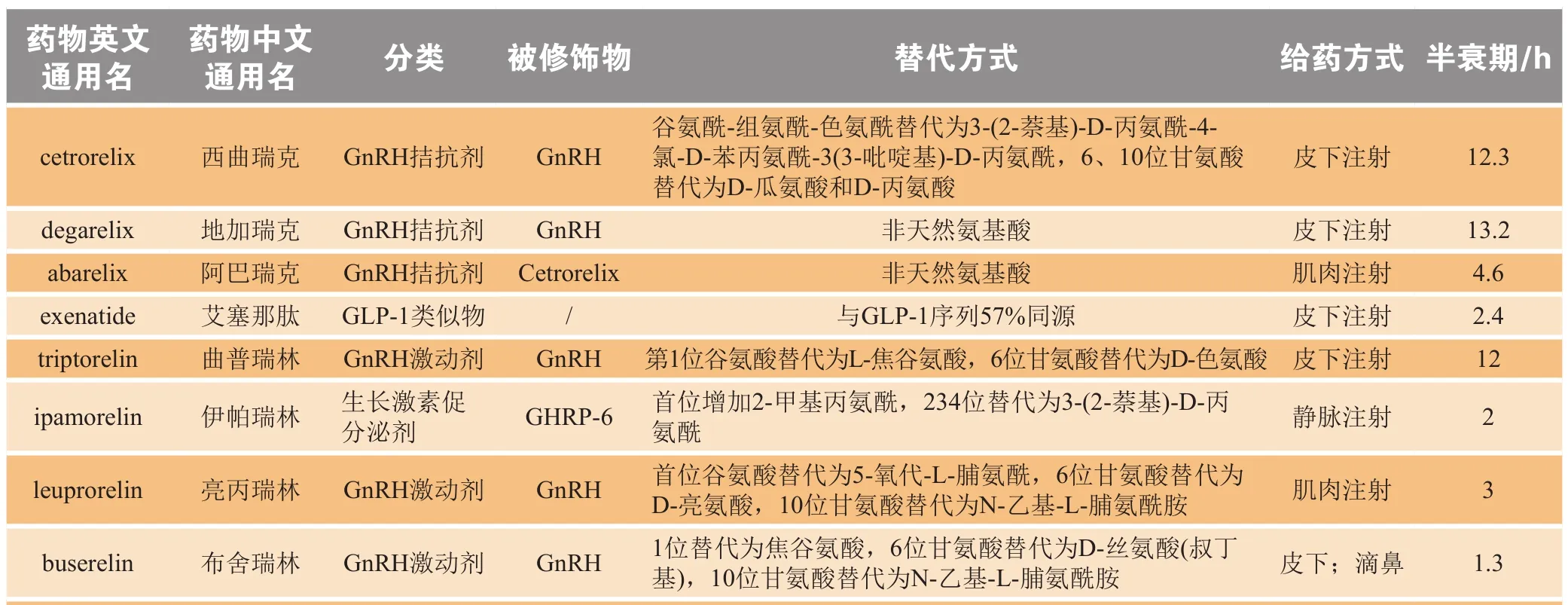
表1 几种通过氨基酸替代延长治疗性多肽半衰期的药物Table 1 Several therapeutic polypeptide drugs with prolonged half-life through amino acid substitutions
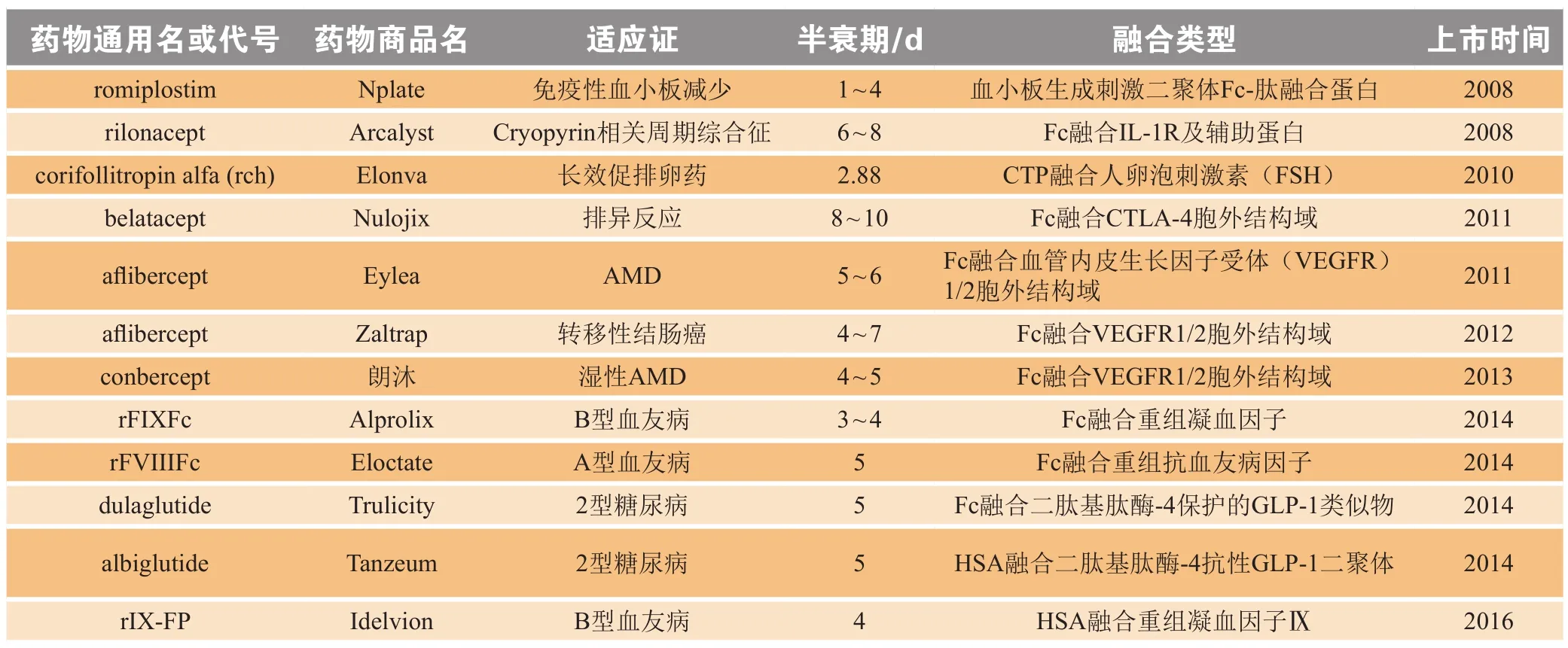
表2 近年获批上市的融合蛋白药物Table 2 Fusion protein drugs approved in recent years

表3 临床在研的融合蛋白药物Table 3 Fusion protein drugs in clinical trials
2.4 糖基化
糖基化通常发生在核酸、蛋白质和脂质等大分子中,在炎症、免疫反应和细胞内转运等生物过程中发挥着重要作用。许多治疗性蛋白多肽类药物在体外进行糖基化后,其药动学和药效学特性可获得改善,此外,使用酶促或化学缀合方法的聚糖修饰还可促进蛋白质靶向疾病影响的组织[28]。研究发现,对重组α-1抗胰蛋白酶(rA1AT)柔性N-末端区域进行N-糖基化修饰后,增加了rA1AT的循环半衰期,同时未改变其蛋白酶抑制活性[29]。特异性血管活性肠多肽受体2激动剂(BAY55-9837)作为2型糖尿病的潜在蛋白质治疗剂,稳定性较差,体内半衰期较短,而将其与壳聚糖修饰的硒纳米颗粒(CS-SeNP)结合后得到的BAY-CS-SeNPs具有良好的稳定性,释放过程持续超过70h,累积释放达到78.9%。此外,CS-SeNP与BAY55-9837缀合后,药物的肾清除率显著降低,且半衰期延长至20.81h[30]。
2.5 环化
线性肽在治疗上受到代谢和构象稳定性差等因素的限制,可能损害其生物活性并影响半衰期,而环肽可以克服这些挑战,因为它们更能抵抗代谢降解且能被设计成所需要的构象。通过环化使多肽类药物获得稳定的刚性结构,是延长血浆半衰期的一种有效途径[31]。环化法能够消除多肽链中带电荷的末端,防止氨基酸的酶解,改善药物代谢稳定性、膜渗透性甚至提高口服生物利用度[32]。奥曲肽是天然生长抑素的环状八聚体合成类似物,其保留了生长抑素的关键Phe-Trp-Lys-Thr部分(含有色氨酸残基),与生长抑素药理作用相似,但作用持续时间更长,在很大程度上延长了体内消除半衰期[33]。普卡那肽(plecanatide)是含有16个氨基酸的环状多肽,2017年经FDA批准上市,商品名为Trulance,给药方式为口服,用于治疗成人慢性特发性便秘[34]。目前,鉴于环化法的优点,此种方法已被广泛应用于制备口服肠道多肽药物,提高药物的生物利用度[35]。环状肽还可与具有高亲和力和选择性的蛋白质靶标结合,用于诊断或治疗,目前已从天然来源如细菌、真菌、植物和动物中分离出针对多种蛋白质靶标的环肽配体[36]。
2.6 与聚合物缀合
2.6.1 PEG化学修饰化学修饰是蛋白多肽类药物常用的一种长效化技术,修饰的常用方法包括N-乙酰化和C-酰胺化[37],氨基酸链中氨基和/或羧基末端的化学修饰可以增强敏感蛋白多肽对蛋白水解酶的稳定性。例如BAX855[PEG-重组凝血因子Ⅷ(rFVIII)],rFVIII经聚乙二醇修饰后,可以减少注射频次,同时也保留了FVIII分子的功效[38]。TP508是一种合成衍生的组织修复肽,在治疗糖尿病足溃疡的Ⅰ/Ⅱ期临床试验中表现出较好的安全性和潜在的功效,但TP508体内半衰期短,研究人员尝试将各种大小的聚乙二醇与TP508的N-末端或内部半胱氨酸上进行共价连接,评估后发现,PEG30k-TP508血浆半衰期相比于TP508延长约19倍,同时还显示出增强的生物活性[39]。对原药N端氨基进行修饰目前已被广泛应用于多肽蛋白类药物的商业化长效技术,例如PEG化粒细胞集落刺激因子(PEGG-CSF)和PEG化白细胞介素-10(PEG-IL-10)早在2006年已申请美国专利,目前也已经有多种PEG化药物被批准上市(见表4)。
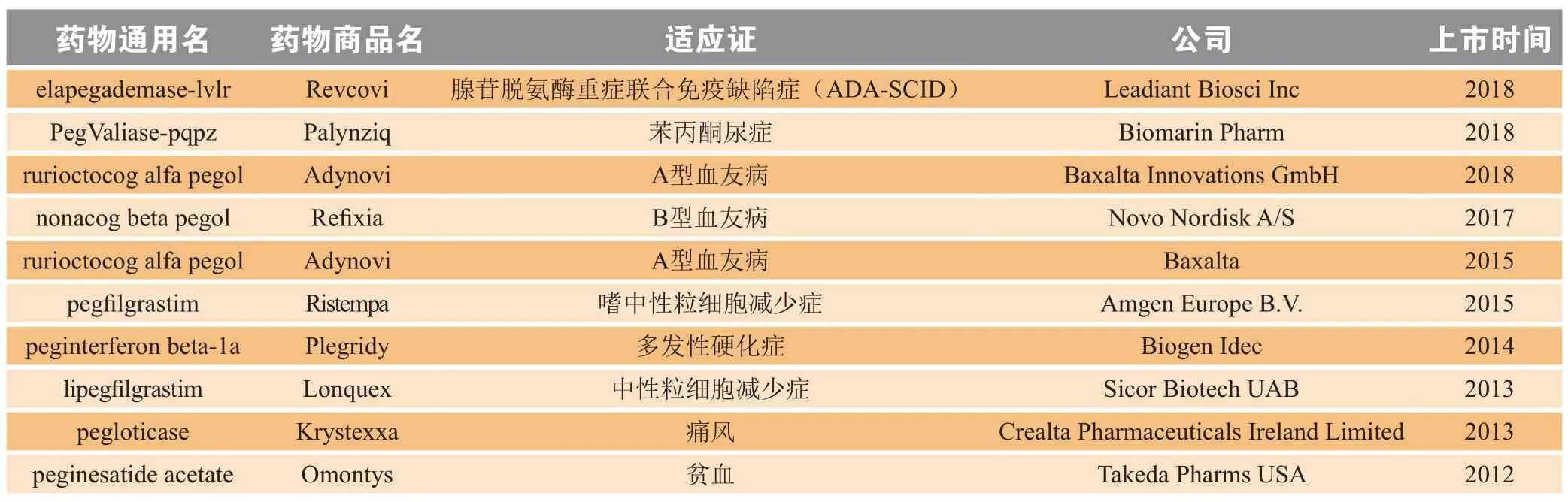
表4 近年来上市的PEG化长效蛋白多肽药物Table 4 Recently approved long-acting PEGylated protein/peptide drugs
2.6.2 非结构化可生物降解蛋白缀合除PEG外,非结构化可生物降解蛋白(XTEN)修饰也能够延长治疗性蛋白多肽的半衰期。XTEN是非结构化的非重复蛋白质聚合物,目标治疗性分子可以与XTEN化学缀合,旨在通过引入类似于PEG的膨胀效应来延长药物的体内半衰期。例如,研究人员将一种抗逆转录病毒肽T-20(用于治疗具有多药耐药性的HIV-1)与XTEN结合,发现其消除半衰期为(55.7±17.7)h,几乎是T-20半衰期的 20倍[40]。
2.6.3 PAS缀合开发出具有扩展的流体动力学体积的构象无序多肽链也是一种延长半衰期的方法,这种无序多肽链包含脯氨酸、丙氨酸和丝氨酸,简称为PAS无序多肽链。PAS序列是亲水的、不带电荷的生物聚合物,在水性缓冲液中形成无规则卷曲结构,其生物物理特性与PEG非常相似,可以通过化学偶联与具有生物活性的蛋白质或多肽缀合,延长其血浆半衰期。此外,PAS可生物降解,避免了在器官中的积聚,在血清中较为稳定,且在小鼠体内很难产生毒性或免疫原性。PAS化已成功应用于多种生物制剂包括细胞因子、生长因子、抗体片段、酶以及各种多肽,其优势在小鼠、猴等各种动物模型中均得到验证[41]。
2.6.4 羟乙基化淀粉缀合阿那白滞素(Anakinra)是人IL-1受体拮抗剂,用于治疗类风湿性关节炎,但此药半衰期较短,临床应用上需要每天注射,极大地降低了患者的顺应性,羟乙基化淀粉(HES)是一种淀粉聚合物,将Anakinra与HES缀合后,得到的复合物稳定性增加,且药动学结果显示其体内半衰期增加6.5倍,AUC增加65倍[42]。
2.7 钉合肽
钉合肽是近年来新出现的一种结构肽,因其结构十分稳定而受到研究者的关注。钉合肽是将肽“钉”成α-螺旋形状,获得优化的交联化学结构,通过交联“装订”赋予肽对蛋白水解降解的抗性并增强其半衰期。钉合肽代表了一种有前途的新方法,可用于调节细胞内蛋白质-蛋白质或蛋白质-DNA相互作用,并提供了瞄准细胞内靶标的机会。LaBelle等[43]基于BIM BH3螺旋模型设计了碳氢化合物钉合肽,其可广泛靶向Bcl-2家族蛋白,从而触发促凋亡活性。ALRN-5281是第一个长效人生长激素释放激素(GHRH)激动剂钉合肽药物,自2013年1月开始进入Ⅰ期临床试验[44]。研究证明,碳氢化合物钉合α-螺旋肽已成为一类新的肽治疗剂[45-46],此类钉合肽具有稳定的构象,能够提高药物的体内生物利用度[47]。
2.8 药物传递系统开发
注射剂进入体内后起效快、体内分布迅速、生物利用度高,但面临着患者依从性差、给药不方便、生产成本高以及使用风险高的问题,因此,研究者也在致力于开发注射剂药物传递系统(DDS),以期将药物在一定时间内递送到特定部位发挥疗效,这种系统能够通过制剂的手段提高药物靶向功效、降低药物的毒副作用[48-49]。Zoladex是已上市多肽植入剂的代表,活性成分是醋酸戈舍瑞林,用于治疗前列腺癌及乳腺癌,可直接注射于皮下或肌内,与其他类型的植入剂相比,无需通过手术取出基质,聚乳酸-羟基乙酸共聚物(PLGA)可自行在体内降解,从而减少了药物不良反应。采用PLGA或聚乳酸(PLA)为骨架材料,包裹药物制成注射微球,在体内也可达到缓释的目的,并且PLGA和PLA是经美国FDA批准的生物材料,可在体内降解。第1个多肽注射微球产品是曲普瑞林PLGA微球,由法国Ipsen公司开发,于1986年上市,可缓释1个月,近年来也有多种长效化缓释微球被批准上市(见表5)。脂质双层和细胞膜结构相似,因此脂质体可以有效地渗透细胞膜以释放药物,而且脂质体可生物降解、基本无毒,可以包封亲水和疏水材料,在药物递送系统中用作药物载体,体内相容性和安全性均较好。例如米伐木肽脂质体注射剂,于2009年上市,给药周期为每周2次,治疗3个月,随后每周1次,治疗6个月,用于治疗非转移性可切除的骨肉瘤。
3 结语
目前,越来越多的研究者将目光转向蛋白多肽类药物,此类药物的研发和上市也呈现出逐步加速的趋势,占据的市场份额也越来越大。一方面,大约80%的蛋白多肽药通过注射途径给药,另一方面,作为长效化技术手段,PEG化和融合蛋白技术在蛋白多肽类药物中的应用较为成熟和普遍,而随着生物技术的发展,开发其他剂型及给药途径也日益受到关注,如DDS。Midasol Therapeutics公司利用胰岛素糖基化纳米粒子开发的经口腔递送系统,以及ActoGeniX公司开发的TopAct技术平台,以期用口服给药替代注射给药途径。研究人员开发了各类药物传递系统,目的是通过载体控制药物的释放、提高药物的稳定性,另外还能提高难溶药物的溶解性、靶向性。目前开发的DDS主要有植入剂、微球注射剂、脂质体注射剂、纳米粒注射剂等,预示着未来缓释、控释和纳米制剂等有非常好的前景。可以相信,未来人们还会不断寻求新的长效化手段,研发出活性更好且更加长效的蛋白多肽类药物,为现有长效技术锦上添花,为广大患者造福。
