超声活化过硫酸盐氧化降解水溶液中的全氟辛烷磺酸盐
2019-04-22李炳智
李炳智,朱 江
(上海市环境科学研究院,上海 200233)
全氟辛烷磺酸及其盐类(PFOS)是一类新型人工合成的持久性有机污染物(POPs),以其稳定的化学结构及疏水疏油的特点而被广泛应用于轻水泡沫灭火剂、电镀铬雾抑制剂、农药生产以及油田回采处理剂等生产领域[1]。从20世纪50年代以来,由于PFOS产品的大量生产、使用和生物富集作用,PFOS已呈现出全球分布的态势。研究证实,在一些环境介质,如地表水[2-3]、土壤[2]、大气[3]、沉积物[3-4]、地下水[2]、污水处理厂[5],甚至鱼类[6]、人体血液[3]和头发[7]内均检测出PFOS的存在,它可通过食物和饮用水摄入的方式进入到人体中[2]。鉴于PFOS分布广、在生态系统中毒性累积性强、难于在环境中降解等独特性,因此已成为国际上水处理领域继内分泌干扰物、持久性有机污染物后又一研究热点。
我国是目前少数生产PFOS类物质的国家之一,也是全球最大的生产国和使用国。我国PFOS生产和使用主要集中在工业发达、新兴产业密集的东部沿海地区[8]。据统计,2010年我国PFOS产量仍有100余t,使用量约80万t[9],PFOS工业源的排放量约为70 t·a-1(占总量70%),排放密度呈现由东向西递减的趋势,经济和工业发展程度最高的京津沪、江苏、广东、浙江和山东是PFOS工业源以及生活源排放密度最高的区域[10]。目前我国水环境监测中普遍检出PFOS,其质量浓度范围介于未检出~458 ng·L-1,局部地区PFOS污染水平超过国外[9,11];而且,国内外尚未制订水环境中PFOS的质量基准/标准。因此,开展我国水环境PFOS的有效降解及其机理研究对于应对全球性PFOS污染,确保我国水环境安全具有重要意义。
PFOS结构中氟具有高的电负性(-4.0),碳原子与之结合形成了稳定的C-F键,键能可达484 kJ·mol-1[12],常规的生物、化学处理不能将其有效去除和矿化[13-14]。近年来,利用高级氧化和还原技术降解PFOS的研究论文数量呈现稳步上升的趋势,这说明学术界对水环境PFOS污染防治领域给予高度关注,且取得了良好的降解效果[15-16]。这类技术以不同的方式产生强氧化性、非选择性的自由基(包括·OH、O-2··和·等),以及高活性、非选择性还原物质或自由基(水合电子、H·和SO-3·等)为特征,产生的自由基随即进攻PFOS,使其得到快速分解。针对污染场地地下水环境中普遍存在的1,1,1-三氯乙烷和共存的溶剂稳定剂1,4-二 口恶烷,笔者所在的课题组成功通过优化超声活化过硫酸盐工艺参数,使二者达到快速同时去除[17-18]。
鉴于此,本文以新型PFOS为模式污染物,利用构建的超声活化过硫酸盐氧化体系,尝试开展该体系降解水中PFOS的降解影响因素及降解产物研究,考察不同反应体系、超声频率、过硫酸盐投量、初始pH值和空化气体种类等对PFOS降解速率的影响,并对PFOS降解过程中产生的自由基及其降解中间产物进行捕捉和鉴定,以期为PFOS污染水环境的治理和修复提供技术支撑。
1 材料和方法
1.1 试验试剂
如无特殊说明,实验中所用化学试剂均为分析纯,未做进一步纯化处理。全氟辛烷磺酸钾(纯度98%,阿拉丁试剂有限公司)、全氟羧酸类中间产物标准品(纯度≥98.0%,上海甄准生物科技有限公司)、过硫酸钠(纯度≥98.0%,国药集团化学试剂有限公司)、5,5-二甲基-1-吡咯-N氧化物(DMPO,97%,阿拉丁试剂有限公司)、乙酸铵(98%)、甲醇(≥99.9%)、硝酸(HNO3)和氢氧化钠(NaOH)均为分析纯试剂,试验用水均采用蒸馏水。HLB(Oasis)固相萃取柱(200 mg/6 mL)购自美国Suplco公司。
1.2 试验方法
试验系统由系列超声波发生器(20、100、200 kHz和400 kHz,输出电功率为0~100 W,声能密度2.67 W·cm-2,中科院东海声学研究所)、超声换能器、40 mL吹扫反应瓶和SDC-6型恒温水槽(江苏佳美仪器制造有限公司)组成。整个系统温度控制在(30±0.2)℃,反应在有聚四氟乙烯膜衬垫盖的玻璃瓶内进行。试验开始前,先将混合均匀的含有一定体积过硫酸盐母液和PFOS母液的溶液注入一系列玻璃瓶内,注入空化气体(氩气、氮气、氧气和空气)20 min,立即旋紧瓶盖,置于换能器上。开启超声波发生器,反应开始计时,超声辐射一段时间后,取样测定水溶液中PFOS浓度及降解中间产物等项目。
1.3 分析方法
PFOS及其降解中间产物分析采用固相萃取前处理-超高效液相-质谱(Agilent 6460,配1260液相系统及工作站)分析。色谱柱ACQUITY UPLC BEH C18column(1.7 μm ×2.1 mm ×50 mm),流动相2 mmol·L-1乙酸铵5%甲醇溶液(A)和甲醇(B)的混合液,采用梯度洗脱模式:0~0.5 min,25%B;5.0 min,85%B;5.1~7.0 min,99%B;7.0~9.0 min,99%B匀速下降至25%B;9.0~12.0 min,25%B。流速 0.4~0.55 mL·min-1,柱温50℃,进样量10 μL,电离源为电喷雾电离源负源(ESI),毛细管电压2.5 kV,离子源温度120℃,脱气溶剂温度350℃,扫描模式MRM,碰撞能量7.0~45.0 eV,锥孔电压15.0~70.0 V。PFOS直接进样检测,PFOS 直接进样检测线性范围为 0.1 μg·L-1~10 mg·L-1,最低检测限为 0.1 μg·L-1。而降解中间产物的分析采用固相萃取前处理后再进样。净化程序如下:分别以5 mL甲醇和超纯水活化HLB固相萃取小柱;取100 mL水样调pH为6,以1.0 mL·min-1流经活化后的小柱;用5 mL 20%甲醇水做淋洗液,氮吹,定容至1 mL。本方法对11种全氟化合物水样的最低检测限介于0.011~0.089 pg·mL-1,固相萃取的平均加标回收率范围为72.6%~144.7%,相对标准偏差介于1.2%~9.2%之间。
以DMPO为自由基诱捕剂,采用德国Bruker EMX-8/2.7型电子顺磁波谱仪半定量分析反应体系产生DMPO-自由基加合物信号。测试条件为:共振频率9.77 GHz,微波功率20.02 mW,调制频率100 kHz,调制振幅1.0 G,扫探宽度200 G,时间常数40.96 ms,扫描时间83.87 s,接收增益为10 000。
氟离子浓度采用美国戴安公司ICS-3000型离子色谱仪进行测定,采用IonPac AS15阴离子交换分析柱(4.0 mm×250 mm),IonPac AG15保护柱(4.0 mm×50 mm),内置数字化电导检测器,KOH淋洗液,梯度淋洗模式:0~17.0 min,3 mmol·L-1;17.1~27.0 min,60 mmol·L-1;27.1~31.0 min,3 mmol·L-1,流速 1.2 mL·min-1,进样体积200 μL。
2 结果与讨论
2.1 超声活化过硫酸盐体系降解PFOS的效果
预试验首先考察了超声活化过硫酸盐体系(US+PS)降解PFOS的效果,并与空白(Control)、单独过硫酸盐氧化(PS)以及单独超声处理(US)的效果进行了比较。试验条件如下:PFOS初始浓度约为10 mg·L-1,过硫酸盐与PFOS摩尔比为50∶1,初始溶液pH值为7.0,温度30℃,超声频率为400 kHz,功率100 W和超声密度2.67 W·cm-2,反应时间设为8 h。PFOS降解试验结果和拟合结果分别如图1和表1所示。
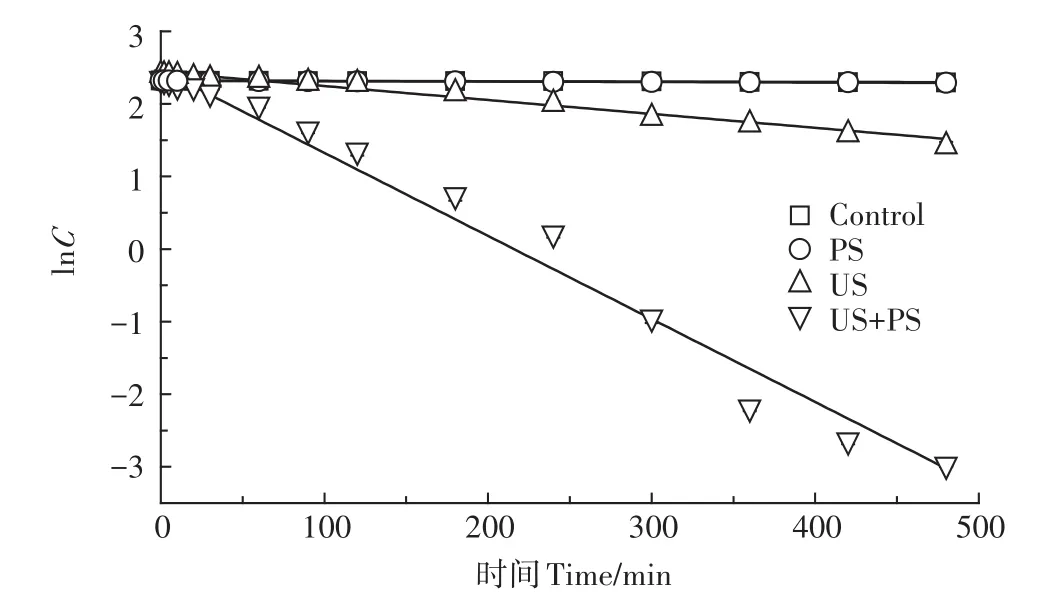
图1 PFOS在不同反应体系降解的浓度对数-时间曲线Figure 1 Logarithm concentration versus time curve of PFOS degradation at different systems

表1 不同体系中PFOS降解的表观准一级反应速率常数值Table 1 Pseudo-first order reaction rate constants of PFOS at different systems
由图1和表1可知,PFOS在4个体系中的降解较符合表观准一级反应动力学模型:ln(Ct/C0)=-kt,R2≥0.941。在Control和PS体系中PFOS几乎无任何降解,在整个反应时间范围内最大降解率低于3%;相比之下,在过硫酸盐未投加情况下,反应速率亦较低,8 h内PFOS降解率仅为62.7%;然而,当超声体系投加过硫酸盐后,8 h内PFOS去除效果显著改善,达到了最高的降解率(99.5%)。据此推测,超声体系投加过硫酸盐后可能生成了更多数量的高活性自由基(SO-4·、·OH、O-2·等),从而极大加速了PFOS的降解和转化。为了更直观地评价超声活化过硫酸盐体系对PFOS降解的强化效果,这里引入增强因子(R)概念,R定义如式1所示。
R=超声活化过硫酸盐体系PFOS降解速率/(单独过硫酸盐体系PFOS降解速率+单独超声体系PFOS降解速率) (1)
由表1计算得到的超声活化过硫酸盐体系降解PFOS的增强因子为5.77,这表明超声活化过硫酸盐体系存在显著的协同效应。
为了证实有活性自由基参与了超声活化过硫酸盐体系PFOS的降解,采用DMPO作为自由基的诱捕剂,利用DMPO与自由基加成反应生成较稳定的加合物这一特性,通过电子自旋共振波谱技术可半定量捕捉到加合物信号,克服了自由基寿命短暂难以捕获的缺点,从而得到直接的试验证据。利用这一技术,我们获得了单独过硫酸盐体系(PS)、单独超声体系(US)和超声活化过硫酸盐体系(US+PS)3个体系反应10 min时的DMPO-自由基加合信号,如图2所示。
由图2可以清晰地发现,在US体系中得到了DMPO加成物的特征四重分裂峰(实心圆点),峰高比1∶2∶2∶1,其超细常数 αN=14.4 G,αH=15.6 G,g值为2.005 5,这些参数与以前的文献报道相一致[19]。这是DMPO与羟基自由基加合的特征参数,由此表明US体系生成了羟基自由基。而在PS体系中,还得到了DMPO加成物的特征六重分裂峰(实心三角),其超细常数αN=13.2 G,αH=9.6 G,αH=1.48 G和αH=0.78 G,与文献报道[20]的硫酸根自由基与DMPO加合物的特征参数相符合,这表明PS体系中存在硫酸根自由基。在“US+PS”体系中,则出现了这两种自由基的叠加峰,而且从加合物的信号强度来看,显著高于PS和US体系之和,这一现象直接解释了“US+PS”体系存在显著协同效应的原因:正是这些明显增多的硫酸根和羟基自由基导致了PFOS的快速降解。
2.2 超声频率对PFOS降解的影响
已有研究证实PFOS在活化过硫酸盐和高频超声作用下能够降解主要归因于活化过硫酸盐产生的高活性物质以及高频超声诱发的较强化学效应,且其降解均遵从表观准一级反应动力学模型[21-23]。本文选择20、100、200 kHz和400 kHz 4个超声频率进行试验,其他试验条件均为:初始PFOS浓度约为10 mg·L-1,过硫酸盐与PFOS摩尔比为50∶1,初始pH值为7.0,温度30℃。表2给出了4个超声频率下PFOS降解的表观准一级反应动力学速率常数。由表2可知,PFOS在4个不同超声频率的降解行为均遵从表观准一级反应动力学规律。随着超声频率的升高,PFOS降解的表观准一级反应速率逐渐增大,由50 kHz时的8.313×10-4min-1增至400 kHz时的1.144×10-2min-1。
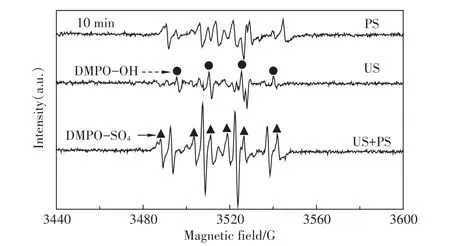
图2 10min时PS、US和US+PS体系DMPO-自由基加合ESR信号Figure 2 ESR spectra of PS,US,and US+PS in the presence of 0.09 mol·L-1DMPO after reaction for 10.0 min
超声频率的影响与空化泡的形成动力学有关,较高超声频率能够实际增加反应体系自由基的数量。较高超声频率时空化泡破灭过程虽不如低频时那样剧烈,但空化过程周期缩短,单位时间内产生的超声空化效应更多,从而更有可能生成更多数量的自由基[24]。由于较高频率时空化泡寿命缩短,自由基在发生自终止反应前有更大几率从空化泡逃逸并迁移至液相本体[25]。
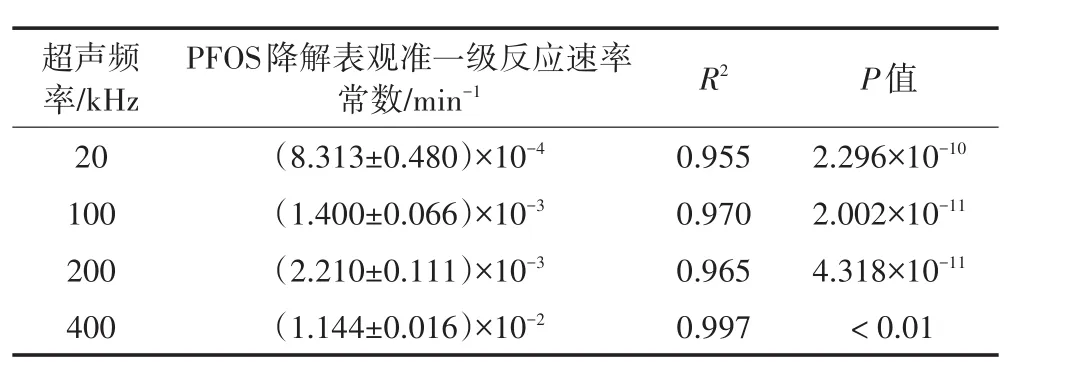
表2 不同超声频率下PFOS降解的表观准一级反应速率常数值Table 2 Pseudo-first order reaction rate constants of PFOS at different ultrasonic frequency
2.3 过硫酸盐浓度对PFOS降解的影响
控制PFOS初始浓度约为10 mg·L-1,初始pH为7.0,温度30℃,超声频率为400 kHz,功率100 W和超声密度2.67 W·cm-2,过硫酸盐投加浓度按与PFOS初始浓度的摩尔比1∶1、5∶1、20∶1、50∶1和100∶1投加,试验研究了过硫酸盐投加浓度对PFOS降解性能的影响。PFOS浓度对数值与时间的拟合试验结果如表3所示。
如表3所示,不同过硫酸盐浓度时PFOS的降解均符合表观准一级反应动力学模型,且与实验数据拟合良好(R2>0.969)。当过硫酸盐与PFOS摩尔比由1∶1增加至50∶1时,PFOS降解的表观准一级反应速率常数由2.470×10-3min-1增至1.144×10-2min-1。然而,当二者摩尔比进一步增大至100∶1时,表观准一级反应速率常数不升反降。
为了探求不同过硫酸盐投量下PFOS降解的表观准一级反应速率常数下降的原因,试验利用ESR测定了不同过硫酸盐投量时DMPO与自由基加合物信号强度的变化,试验结果如图3所示。
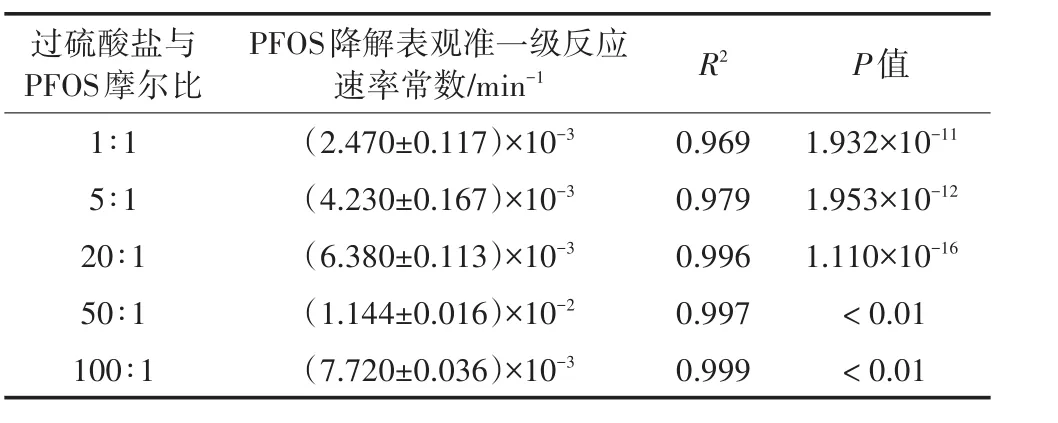
表3 不同过硫酸盐浓度时PFOS降解的表观准一级反应速率常数值Table 3 Pseudo-first order reaction rate constants of PFOS at different persulfate dosages
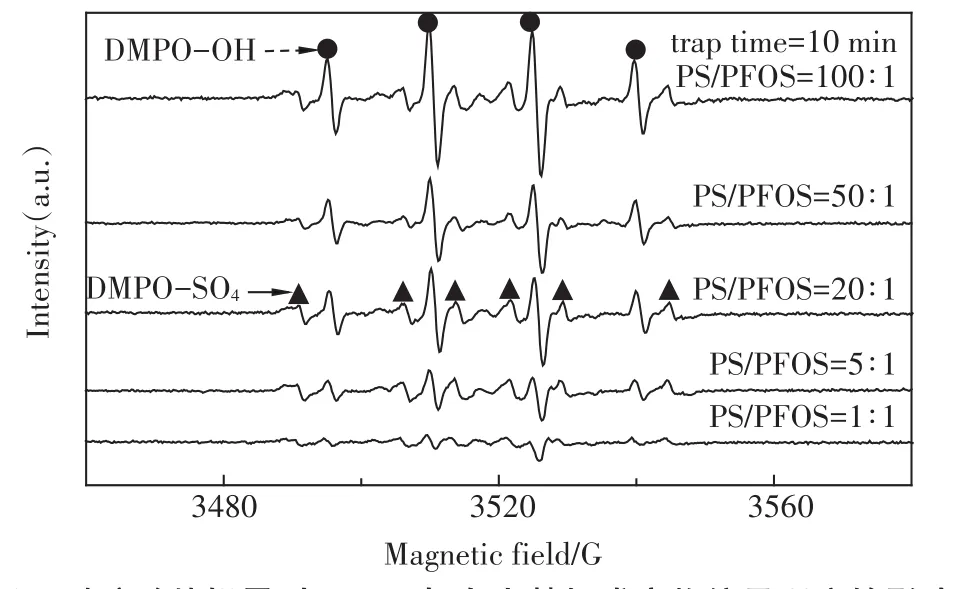
图3 过硫酸盐投量对DMPO与自由基加成产物信号强度的影响Figure 3 Effect of persulfate dosage upon the intensity of the formation of DMPO adducts
由图3可以看出,过硫酸盐与PFOS摩尔比从1∶1增加至100∶1时,DMPO-SO4以及DMPO-OH峰强度和自由基浓度也相应增加,但试验结果中却未出现与此相匹配的PFOS强化降解现象。据此推测,可能的原因有:
(1)硫酸根自由基和羟基自由基一旦生成即随之发生一系列化学反应,包括活性物质的形成、自由基重组以及过硫酸盐猝灭反应,如方程式2~方程式10所示[26]。尽管羟基自由基(E0=+2.7 V)有比硫酸根自由基(E0=+2.6 V)较高的氧化还原电位值[27],但伴随硫酸根自由基生成而产生的二价硫酸根离子却会抑制上述两种自由基的反应活性[28]。


(2)硫酸根自由基自身的重组反应(式5)、羟基自由基自身重组反应(式4)以及二者之间的反应(式8)速率比羟基自由基、硫酸根分别与PFOS的反应以及过硫酸盐的猝灭反应(式9、式10、式6和式7)约高1~3个数量级[16,29-31]。由于自由基之间的竞争性消耗作用,当过硫酸盐投加过量时这些反应会导致自由基的利用率低下。(3)其他也有类似研究表明,对于有机物氧化而言,的确存在一个最佳的过硫酸盐投加值,如高于此临界值,污染物的降解就会受到一定程度的抑制[32-34]。
2.4 反应体系初始pH值对PFOS降解的影响
为了考察初始pH值变化对超声活化过硫酸盐体系降解PFOS速率的影响,控制试验条件为:初始PFOS浓度约10 mg·L-1,初始pH值分别为3.0、7.04和11.03,温度30℃,超声频率400 kHz,功率100 W、超声密度2.67 W·cm-2,过硫酸盐与PFOS初始浓度的摩尔比为50∶1。PFOS浓度对数值与时间的表观准一级反应动力学拟合试验结果如表4所示。
如表4所示,随着溶液初始pH值由3.0增至11.03,相应的PFOS降解的表观准一级反应速率常数由0.013 75 min-1下降至0.009 47 min-1。之所以出现上述试验现象,这可能是因为:(1)酸性条件时,由于酸的催化作用会生成额外多的硫酸根自由基[28];(2)在碱性条件时,硫酸根自由基会与氢氧根离子反应生成更多的羟基自由基,如方程式(3)所示;羟基自由基通常通过H原子抽提进攻有机物分子产生水,但PFOS分子本身不含H原子,无法发生H原子抽提反应,水溶液中二者间的反应活性较差,从而减慢了PFOS的降解速率,故碱性条件不利于硫酸根自由基对PFOS的降解。随着反应的进行,尽管溶液pH值逐渐下降(例如初始pH值分别为3.00、7.00和11.03时,经过近500 min,反应终了pH值逐渐分别降至2.70、6.42和10.38),但是溶液初始pH值越高,氢氧根离子浓度越高,被其消耗的硫酸根自由基也越多,从而削弱了反应体系的氧化作用。

表4 不同初始pH值时PFOS降解的表观准一级反应速率常数值Table 4 Pseudo-first order reaction rate constants of PFOS at different initial pH values
2.5 空化气体种类对PFOS降解的影响
空化气体种类(如氮气、氧气、空气和氩气等)对超声的影响主要表现为反应体系活性物质(如H·、·OH、H2O2、H2、HO2·等)种类及其产率的差异,进而对污染物的降解行为产生影响[35]。4种空化气体对超声活化过硫酸盐体系降解PFOS速率的影响结果,见表5。
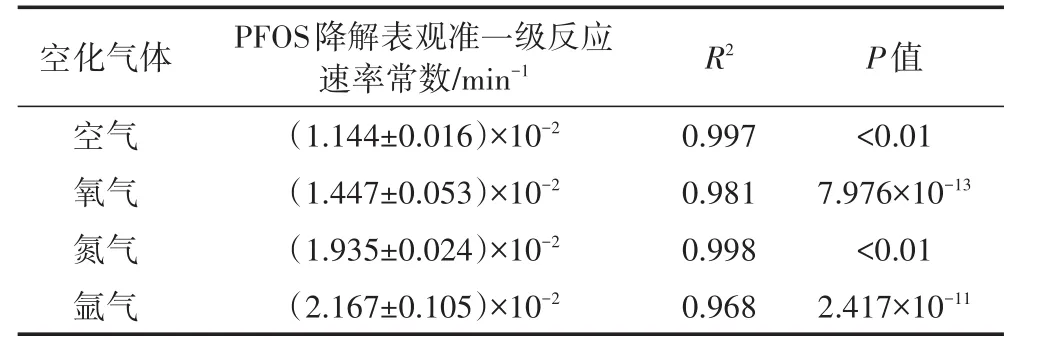
表5 不同空化气体PFOS降解拟合的表观准一级反应速率常数值Table 5 Pseudo-first order reaction rate constants of PFOS at different cavitation gas
由表5可知,4种空化气体(氮气、氧气、空气和氩气)存在下,PFOS降解均遵从表观准一级反应动力学,反应速率常数大小顺序为:空气<氧气<氮气<氩气。之所以出现上述结果,其原因可能是:(1)比热大的空化气体更有利于空化气泡的崩溃,由空化效应获得的声化学效应越大,单原子气体比双原子气体、杂原子气体更适合作为空化过程的气源[36];(2)空化气体导热率越大,空化气泡崩溃过程积累热量将更多地传给周围液体,从而降低最高温度值;(3)溶解度大的空化气体会降低空化阈值和空化强度[37]。根据上述试验结果可知,PFOS降解效果最佳的空化气体为氩气。
目前,虽然高级氧化(臭氧氧化、电化学氧化、超声、过硫酸盐氧化和光解)和还原工艺(零价铁和柠檬酸钛)处理水中PFOS的研究报道较多,但由于这些研究中PFOS初始浓度范围设置较为宽泛,使得很难客观地比较这些方法在降解PFOS性能上的优劣[16]。文献报道中单独超声(200 kHz,200 W)和50℃热活化过硫酸盐分别降解 20 μmol·L-1的PFOS,其一级反应速率常数分别为0.016 min-1和0.000 47 min-1[16],与本文中PFOS降解速率常数基本相当。高效超声换能器研发成功之前,综合过硫酸盐和超声两种AOPs工艺的耦合协同作用提高对PFOS的降解效率,起到“1+1>2”增效作用的同时显著降低了超声能耗。
2.6 降解中间产物鉴定及可能的降解机理
为了深入研究PFOS在超声活化过硫酸盐体系中的降解机理,试验采集了不同反应时间间隔内的PFOS溶液样品,并将其混合。利用超高效液相色谱-质谱仪(UPLC-MS)捕捉到了一系列PFOS降解产物,其典型色谱图如图4所示。
根据与全氟代标准物质色谱图比对,确认检测到的8种物质及停留时间(Rt)分别为:全氟乙酸(Rt=1.22 min)、全氟丙酸(Rt=1.27 min)、全氟丁酸(Rt=2.49 min)、全氟戊酸(Rt=3.13 min)、全氟己酸(Rt=3.73 min)、全氟庚酸(Rt=3.79 min)、全氟辛酸(Rt=4.18 min)、未知物质(Rt=4.45 min)和PFOS(Rt=4.55 min)。
有关超声和活化过硫酸技术降解PFOS产物研究数量相当有限。日本大阪府立大学Moriwaki等[38]利用200 kHz超声降解PFOS的产物包括:三氟乙酸、五氟丙酸、七氟丁酸、九氟戊酸、全氟己酸、全氟庚酸、全氟己烷磺酸和全氟辛酸等;杨佘维等[39]发现,真空紫外耦合高频超声体系中PFOS降解中间产物为7种短链全氟羧酸(三氟乙酸、五氟丙酸、七氟丁酸、九氟戊酸、全氟己酸、全氟庚酸和全氟辛酸)以及3种短链全氟磺酸(全氟庚烷磺酸、全氟己烷磺酸和全氟丁烷磺酸)。由此可见,文献报道的PFOS降解产物与本文研究结果基本一致,即PFOS在反应体系中主要分解为两类物质:短链的全氟代羧酸类和全氟代磺酸类中间产物。
超声活化过硫酸盐体系降解有机化合物主要遵从热解和(或)自由基(如SO-4·、·OH等)诱导的反应机制。PFOS较高的辛醇-水分配系数、亨利系数和蒸汽压使其具有较好的亲水性和较低的挥发性,因此可以预知:反应初始阶段其可能在空化泡内或空化泡-溶液交界面处进行热解反应,以及在液相本体中进行自由基的氧化反应[40]。结合分析测定的中间产物,超声活化过硫酸盐体系降解PFOS可能的机理见图5。标框的物质为本研究检测到的中间产物。
文献报道PFOS钾盐的pKa值为-3.5[41],在所有可能的环境条件(强酸性条件除外)下,PFOS钾盐均以离子状态存在。根据检测到的中间产物和相关文献报道,推测可能的反应机理有以下两种途径:(1)水相本体反应。在超声场和硫酸根自由基的诱导下,PFOS阴离子首先被提取一个电子,形成C8F17SO3·自由基,然后脱去磺酸基,生成C8F17·自由基。C8F17·自由基末端的碳原子具有较高的电荷密度,极易与氧化剂反应生成热不稳定全氟代醇类物质C8F17OH;C8F17OH脱去氟化氢而生成酰基氟C7F15COF,C7F15COF极易水解,生成少1个CF2单元的C7F15COOH。然后SO-4·自由基进攻C7F15COO-阴离子,转化为硫酸盐、二氧化碳和C7F15·;C7F15·重复上述成醇、脱氟化氢和水解步骤,短链的C2~C7全氟羧酸降解产物会依次产生,最终PFOS逐步降解成氟离子和二氧化碳。(2)气泡-水界面反应。首先PFOS被吸附到气泡-水的界面,通过超声产生局部的高温高压效应,PFOS末端的C-S键热解断裂,释放气体SO3至水相,自身转化为气态全氟烯烃;气态全氟烯烃在高温条件下,裂解为氟化物自由基,在自由基和高温的进一步作用下继续转化为二氧化碳、一氧化碳和氟化氢。
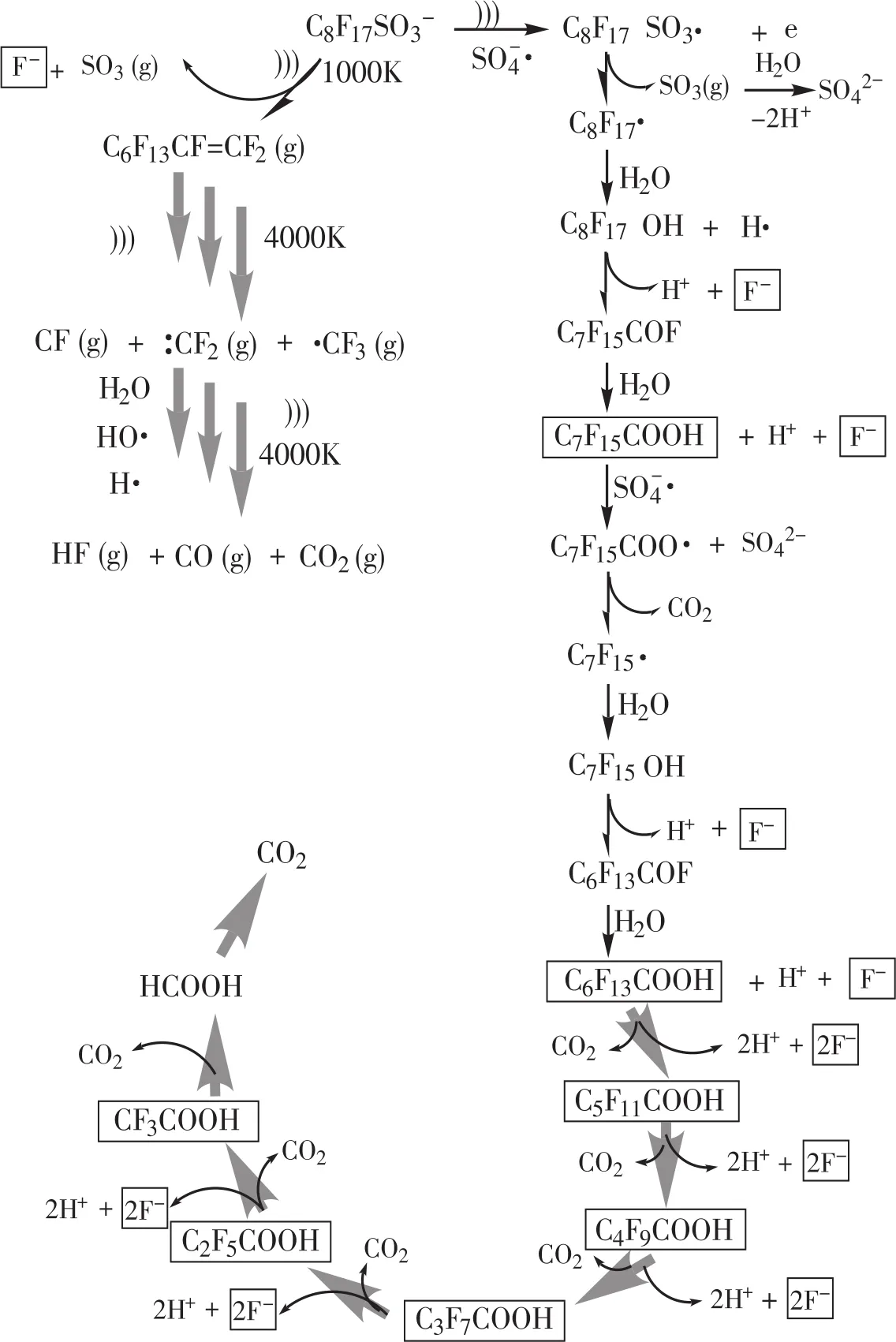
图5 PFOS在超声活化过硫酸盐氧化体系中可能的降解途径Figure 5 Proposed PFOS degradation pathway in the sono-activated persulfate oxidation process
由PFOS的降解途径可知,PFOS的降解主要在气相反应和液相反应作用下逐渐分解为短链的全氟羧酸和全氟磺酸中间产物;然而,有相当多的研究表明,这些中间产物的降解速率要慢于母体污染物PFOS[16]。例如,在本文的试验条件下,PFOS经过8 h基本完全降解,而其脱氟率仅为90%,这说明仍有部分含氟PFOS降解中间产物尚未得到完全脱氟。在PFOS污染水体处理实践中,PFOS母体污染物的完全降解并不是处理的终点;在此之后仍需继续延长处理时间,以使得PFOS降解中间产物(如短链全氟羧酸和全氟磺酸类等)得到彻底降解。
关于世界上其他国家和地区水环境中PFOS的浓度值,已有文献报道。PFOS在地表水、污水处理厂出水和饮用水中浓度分别介于1~7、7~50 ng·L-1和1~100 ng·L-1[42],基本与我国水环境中PFOS的检测浓度相当。为此,一些国家已制定饮用水中全氟化合物健康指导值为0.04~0.5 μg·L-1[43]。本文之所以设置的PFOS模拟浓度较环境介质报道值高,是为了便于检测浓度较低的PFOS降解产物,以更好地阐释其降解机理。后续研究将基于我国环境介质中PFOS检出浓度开展有针对性的PFOS氧化和还原降解的对照研究。
3 结论
(1)与单独超声和单独过硫酸盐氧化体系相比,超声活化过硫酸盐体系能够显著加速PFOS的降解和转化,存在明显的协同效应,增强因子达到5.77,其本质原因在于:协同体系生成了明显增多的·OH和SO-4·。
(2)该体系降解PFOS(初始浓度约10 mg·L-1)较佳的实验条件为:温度30℃、超声频率400 kHz、功率100 W、超声密度2.67 W·cm-2、过硫酸盐与PFOS摩尔比为50∶1,初始pH值为7.0,空化气体为氩气,反应时间为8 h。
(3)根据检测到的7种PFOS降解产物,给出了PFOS在超声活化过硫酸盐体系中可能的降解途径。
