高效液相色谱-蒸发光散射法测定黄氏响声含片中贝母素甲和贝母素乙含量
2019-04-18赵懿清高秋芳郭卫东邹济高
陆 丹,赵懿清 ,高秋芳 ,郭卫东,邹济高
(1.江苏省无锡市第三人民医院,江苏 无锡 214000; 2.江苏省无锡济民可信山禾药业股份有限公司,江苏 无锡 214000)
黄氏响声含片为复方制剂,由薄荷、浙贝母、连翘、蝉蜕、胖大海、酒大黄、川芎、儿茶、桔梗、诃子肉、甘草、薄荷脑等药材组方,可疏风清热、化痰散结、利咽开音。方中浙贝母为君药,可解毒散结消痈[1],其主要成分为贝母素甲(浙贝母碱)和贝母素乙(去氢浙贝母碱)[2]。本研究中采用高效液相色谱-蒸发光散射(HPLC-ELSD)法测定黄氏响声含片中贝母素甲和贝母素乙的含量,为该制剂的质量控制奠定基础。现报道如下。
1 仪器与试药
1.1 仪器
Agilent 1260型高效液相色谱仪,包括OpenLab工作站(美国Agilent公司);ALLTECH 3300型蒸发光散射检测器(美国奥泰科技<中国>有限公司);BP211D型电子分析天平(德国Sartorius公司);KQ250DB型数控超声波清洗器(昆山市超声仪器有限公司,功率为250 W)。
1.2 试药
黄氏响声含片(无锡济民可信山禾药业股份有限 公 司,批号分别为 171112,171121,171124,171204,171206,171207,180201,180204,180301,180303,180401,180403,180405,规格为每片 0.6 g);贝母素甲对照品(批号为110750-201612,纯度为96.2%)、贝母素乙对照品(批号为110751-201712,纯度为97.7%),均购自中国食品药品检定研究院;乙腈、甲醇为色谱纯,其余试剂均为分析纯,水为超纯水。
2 方法与结果
2.1 色谱条件
色谱柱:Phenomenex Gemini-NX C18柱(150 mm×4.6 mm,5 μm);流动相:乙腈 -水(65 ∶35,V/V,含0.035% 二乙胺);流速:1.0 mL/min;柱温:30 ℃;进样量:20 μL;ELSD参数:漂移管温度为70℃,载气流速为1.4 L/min。
2.2 溶液制备
取贝母素甲和贝母素乙对照品各适量,精密称定,置容量瓶中,加流动相溶解并定容,制成贝母素甲和贝母素乙质量浓度分别为0.515 0 g/L和0.503 1 g/L的对照品贮备液,取适量,制得贝母素甲和贝母素乙质量浓度分别为0.506 4 g/L和0.503 7 g/L的混合对照品溶液。取样品适量,捣碎,研细,过4号筛,取约5 g,精密称定,置具塞锥形瓶中,加入浓氨水-乙醇(1∶2,V/V)10 mL,密塞,浸泡 30 min,加乙醚-三氯甲烷-乙醇(50 ∶16 ∶5,V/V/V)混合溶液 25 mL,超声处理 30 min(控制水温低于30℃),滤过,滤渣用适量乙醚-三氯甲烷 -乙醇(50∶16∶5,V/V/V)洗涤 3次,合并滤液和洗液,60℃水浴挥干,残渣用流动相2 mL溶解,经0.22 μm微孔滤膜滤过,取续滤液,即得供试品溶液。按处方和工艺制备缺浙贝母的阴性样品,按供试品溶液制备方法制成阴性对照品溶液。
2.3 方法学考察
系统适用性试验:取2.2项下3种溶液各适量,按拟订色谱条件进样测定,记录色谱图(图1)。结果理论板数按贝母素甲、贝母素乙峰计均不低于3 000,分离度均大于1.5,基线分离良好,保留时间分别为6.273 min和7.957 min,对称因子分别为0.82和0.84。
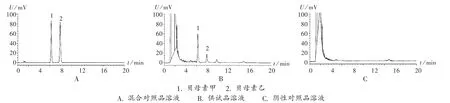
图1 高效液相色谱图
线性关系考察:分别精密量取混合对照品溶液0.5,1.0,2.0,2.5,4.0 mL,置 10 mL 容量瓶中,加流动相定容,摇匀,制得系列对照品溶液,精密量取20 μL,按拟订色谱条件进样测定,记录峰面积。以待测成分质量浓度(μg/mL)的自然对数值(X)为横坐标、峰面积的自然对数值(Y)为纵坐标进行线性回归,得贝母素甲和贝母素乙回归方程分别为Y甲=1.4647X甲-0.1089(r=0.9998,n=5),Y乙=1.453 6X乙-0.016 0(r=0.999 9,n=5)。结果表明,贝母素甲和贝母素乙质量浓度线性范围分别为25.32~202.56 μg/mL和25.19~201.48 μg/mL。
定量限与检测限考察:精密量取混合对照品溶液适量,倍比稀释,并按拟订色谱条件进样测定,以信噪比(S/N)为10∶1和3∶1分别计算定量限、检测限。结果贝母素甲和贝母素乙的定量限分别为189.9ng和251.9ng,检测限分别为79.1 ng和75.6 ng。
精密度试验:取混合对照品溶液适量,按拟订色谱条件连续进样测定6次,记录峰面积。结果贝母素甲和贝母素乙峰面积的RSD分别为1.51%和0.85%(n=6),表明仪器精密度良好。
稳定性试验:取供试品(批号为180403)溶液适量,分别于室温下放置 0,1,2,4,8,12 h,按拟订色谱条件进样测定,记录峰面积。结果贝母素甲和贝母素乙峰面积的RSD分别为2.28%和1.77%(n=6),表明供试品溶液室温放置12 h内基本稳定。
重复性试验:称取样品(批号为180403)适量,精密称定,同法制备供试品溶液,再按拟订色谱条件进样测定,记录峰面积,并计算含量。结果贝母素甲和贝母素乙平均含量分别为 36.619 μg/g和 15.677 μg/g,RSD分别为2.47%和2.24%(n=6),表明方法重复性良好。
加样回收试验:取已知含量样品(批号为180403)6份,每份约2.5 g,精密称定,分别加入一定质量浓度的对照品溶液,同法制备供试品溶液,再按拟订色谱条件进样测定,记录峰面积,并计算回收率。结果见表1。

表1 加样回收试验结果(n=6)
2.4 样品含量测定
取13批样品各适量,同法制备供试品溶液,再按拟订色谱条件进样测定,平行测定3次,记录峰面积,并计算样品含量。结果见表2。可见,各批次间贝母素甲和贝母素乙的含量存在一定差异,平均含量分别为0.030 7 mg/g和0.0136 mg/g,RSD分别为10.02%和9.01%,平均总含量为0.0444mg/g,RSD为8.57%;以平均含量的80%制订含量下限,样品中贝母素甲、贝母素乙及总含量应分别不低于 0.024 6 mg/g,0.010 9 mg/g,0.035 5 mg/g,各批样品均符合要求。
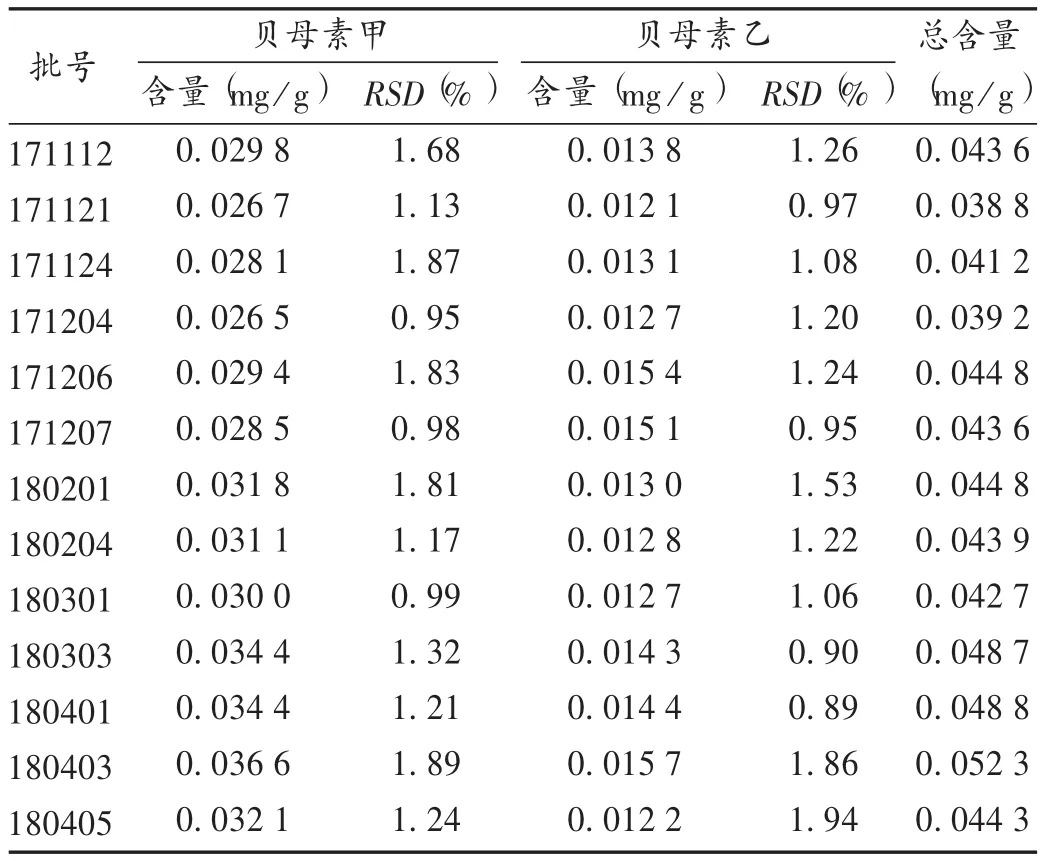
表2 样品含量测定结果(n=3)
3 讨论
3.1 分析条件选择及优化
贝母类生物碱为异甾类生物碱,文献报道的测定方法有薄层扫描法、HPLC法、柱前衍生化HPLC法、柱前衍生化气相色谱法[3]、近红外光谱法[4]、化学发光分析法等[5]。由于贝母素甲无紫外吸收,贝母素乙仅有末端吸收,不宜用紫外检测器检测,同时考虑到灵敏度及操作的简便性,因此采用HPLC-ELSD法分析,效果较佳。
为了降低灵敏度损失,同时使流动相充分挥发,选择不分流模式。在此基础上考察漂移管温度及载气流速,最终确定漂移管温度70℃,载气流速1.4 L/min。
2015年版《中国药典(一部)》浙贝母药材和复方黄氏响声丸项下均采用HPLC-ELSD法测定贝母素甲和贝母素乙的含量,试验中考察了流动相乙腈-水的比例(70 ∶30,65 ∶35)及二乙胺的加入量(0.030%,0.035%,0.050%)对结果的影响。结果显示,流动相为乙腈-水(65∶35,V/V,含 0.035%二乙胺)时,待测成分分离良好[1]。
3.2 样品前处理方法优化 [1,6-15]
以待测成分含量为指标,优化样品前处理方法。考虑峰面积的适宜性,供试品的取样量选择5 g,保持取样量不变,考察浓氨水-乙醇(1∶2,V/V)混合液的用量为 2,3,5,10,15,20 mL 时的浸泡效果。结果显示,当用量为20 mL时色谱峰基线不稳,10 mL和15 mL的浸泡效果均较佳,考虑到经济性,混合液用量选择10 mL。分别用超声和加热回流方式提取供试品溶液,结果两者提取效果基本一致,超声提取更简便,故选择超声提取。比较乙醚 -氯仿 -乙醇(50∶16∶5,V/V/V)、三氯甲烷 -甲醇(4∶1,V/V)和流动相为超声溶剂时的提取效果,结果显示,用流动相处理的样品很黏稠,糖类物质被提取出而影响测定,采用乙醚-氯仿-乙醇(50∶16∶5,V/V/V)作为超声溶剂,提取效果最佳,可满足测定要求。考察超声提取溶剂量分别为15,25,50 mL时的提取效果,结果表明,加入混合提取液25 mL即可将贝母素甲、贝母素乙提取完全。设置超声时间为15,30,45 min,考察提取效果,结果超声处理30 min即可将贝母素甲、贝母素乙提取完全。比较乙腈、水、甲醇、流动相4种复溶溶剂,结果显示,贝母素甲、贝母素乙在水中的溶解性较差,水复溶样品未见出峰,用流动相溶解样品处理效果最佳,可满足测定要求。
综上所述,本研究中建立的方法操作简便、准确,精密度、稳定性、重复性均良好,可用于测定黄氏响声含片中贝母素甲和贝母素乙的含量。
