Ursodeoxycholic acid ameliorates hepatic lipid metabolism in LO2 cells by regulating the AKT/mTOR/SREBP-1 signaling pathway
2019-04-15JieHuWeiHongKanNanYaoXiaoHongZhuZhiYunChenLeiYe
Jie Hu, Wei Hong, Kan-Nan Yao, Xiao-Hong Zhu, Zhi-Yun Chen, Lei Ye
Jie Hu, Xiao-Hong Zhu, Lei Ye, Department of Infectious Diseases, The First Affiliated Hospital of Zhejiang Chinese Medical University, Hangzhou 310006, Zhejiang Province, China
Wei Hong, Kan-Nan Yao, Zhi-Yun Chen, the Second Central Laboratory, The First Affiliated Hospital of Zhejiang Chinese Medical University, Hangzhou 310006, Zhejiang Province,China
Abstract BACKGROUND Nonalcoholic fatty liver disease (NAFLD), the most common chronic liver disease, can progress into nonalcoholic steatohepatitis (NASH), cirrhosis, and even hepatocellular carcinoma. Bile acids such as ursodeoxycholic acid (UDCA)play an essential role in the pathogenesis of NAFLD by regulating the level of sterol regulatory element-binding protein (SREBP) 1c, but the underlying regulatory mechanism remains elusive. Increased evidence indicates that the AKT/mTOR/SREBP-1 signaling pathway is a key pathway to regulate hepatic cellular lipid metabolism. UDCA may regulate the AKT/mTOR/SREBP-1 signaling pathway to ameliorate hepatic lipid metabolism.AIM To investigate the functional mechanism of UDCA in an oleic acid (OA)-induced cellular model of NAFLD.METHODS The cellular model of NAFLD was established using OA and treated with UDCA.First, the best concentration of UDCA was selected. For the best time-dependent assay, cells were stimulated with OA only or co-treated with OA and 2 mmol/L UDCA for 24 h, 48 h, and 72 h. Oil red O staining was used to observe the accumulation of intracellular lipids, while the intracellular contents of triglyceride, alanine aminotransferase (ALT), gamma-glutamyl transpeptidase(GGT), and aspartate aminotransferase (AST) were detected by enzymatic methods. Meanwhile, the expression levels of AKT/mTOR/SREBP-1 signaling pathway-related proteins were detected by real-time PCR and Western blot.RESULTS In the NAFLD cell model established with LO2 cells induced using OA, lipid accumulation was obvious. UDCA significantly inhibited lipid accumulation at different concentrations (especially 2 mmol/L) and decreased cell growth ability at different time points. The biochemical parameters like ALT, AST, and GGT were significant improved by UDCA. UDCA treatment vividly repressed the activation of AKT, mTOR, and CRTC2 and the expression of nSREBP-1 in LO2 cells induced with OA.CONCLUSION Our findings demonstrate the effect of UDCA in improving NAFLD. UDCA attenuates OA-induced hepatic steatosis mainly by regulation of AKT/mTOR/SREBP-1 signal transduction.
Key words: Ursodeoxycholic acid; Hepatic lipid metabolism; AKT/mTOR/SREBP-1;Hepatic steatosis
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is characterized by macrovesicular fat accumulation of more than 5% in the hepatocytes of patients, due to the deposition of fat for reasons other than excessive alcohol use[1]. The prevalence of NAFLD ranges from 9%to 36.9% in different regions of the world, and is growing rapidly worldwide[2,3]. In some cases, the progression of NAFLD may lead to the formation of nonalcoholic steatohepatitis (NASH) or other types of liver diseases[4]. Hepatic lipid metabolism dysbiosis-induced hepatic steatosis is regarded as the major cause of NAFLD[5]. Thus, investigation of the proposed mechanisms of hepatic lipid metabolism in the pathogenesis of NAFLD is potentially contributing to improving the pathological symptoms of NAFLD and NAFLD-related liver disease.
As amphipathic molecules, bile acids (BAs) are generated from cholesterol oxidation in the liver and play an essential role in the progression of NAFLD[6,7]. At the right concentration, BAs function as a master regulator in the digestion and absorption of lipids. However, excessive BAs exert detrimental effects on hepatocytes[8]. BAs can maintain triglyceride (TG) homeostasis and act as a metabolic regulator of glucose uptake and lipid metabolism[9]. It is confirmed that BAs binding to activated farnesoid X receptor (FXR) in the liver inhibit sterol regulatory elementbinding protein 1c (SREBP1c)-mediated lipogenesis[10,11]. Conversely, activation of FXR also suppresses synthesis of BAs when BAs are excessive[12]. The production of BAs is the main route of cholesterol catabolism and disturbance of the enterohepatic circulation of bile acid leads to the decrease of endogenous cholesterol[3].Downregulation of endogenous cholesterol induces the generation of mature SREBP-1 which can initiate lipogenesis in turn. Thus, the complicated correlation of BAs and hepatic lipid metabolism is essential for the maintenance of liver homeostasis.
Ursodeoxycholic acid (UDCA) is one of the secondary BAs and has been widely used for the treatment of cholestatic liver diseases, such as gallstones, and nonsurgical treatment of primary biliary cirrhosis, through its cytoprotective effect and anti-apoptotic activities in the liver[13,14]. UDCA-mediated glutathione synthesis through the activation of the PI3K/AKT/Nrf2 pathway exerts antioxidative action and is regarded as a potential treatment for chronic hepatitis[15]. Additionally, it has been reported that UDCA administration promotes the progression of lipogenesis in the livers of obese people by the antagonism of FXR[16]. In contrast, recent studies have indicated that UDCA and taurine-conjugated UDCA facilitate the improvement of abnormal glucose metabolism and insulin resistance[17]. UDCA also acts as a protection factor for hepatic steatosis and hepatitis in mice[18]. Although application studies of UDCA in NAFLD have been widely used and have achieved good results,the underlying mechanism of UDCA in regulating the pathogenesis of NAFLD remains largely unknown.
Herein, we discovered that UDCA administration reduced oleic acid (OA)-induced production of lipid droplets and disturbance of hepatic lipid metabolism by regulating the AKT/mTOR/SREBP-1 signaling pathway. These findings could possibly provide a great reference for clinical application of UDCA in NAFLD.
MATERIALS AND METHODS
Cell culture and treatment
Human LO2 cells were purchased from Bogoo Biotechnology (Shanghai, China). Cells were cultured in RPMI 1640 medium (HyClone) containing 10% fetal bovine serum(FBS; Wisent) and incubated at 37 °C in a humidified atmosphere containing 50 mL/L CO2. An in vitrocell model of NAFLD was established by treating cells with 20 μg/mL OA for 48 h. In cell viability experiments, cells were treated with OA only or cotreated with OA and a concentration gradient of UDCA for 48 h. For time-dependent assays, cells were stimulated with OA only or co-treated with OA and 2 mmol/L UDCA for 24 h, 48 h, and 72 h. For the detection of the lipid profile and the involved signaling pathways, cells were incubated with OA only or co-treated with OA and 2 mmol/L UDCA for 72 h.
Oil red O staining
After treatment with OA or UDCA, cells were washed twice with PBS and fixed using 10% paraformaldehyde overnight. Then, cells were washed twice again and stained in oil red O solution (isopropanol: 0.5% oil red O, 3:2) for 20 min at room temperature.Oil red O solution was removed, and the cells were stained with haematoxylin for 30 s. Next, cells were differentiated using hydrochloric acid alcohol (1:1) for 5-10 s. After washing again, oil red O staining results were observed under an optical microscope.MTT assay
LO2 cells were seeded in 100 μL in a 96-well plate at a density of 5 × 104cells/mL and incubated at 37 °C in a humidified atmosphere containing 50 mL/L CO2overnight.Then, cells were treated with OA and UDCA according to the method we described above. At different time points, 100 μL MTT (1 mg/mL) was added into the wells after removing the medium. After incubation for another 4 h, 150 μL DMSO was used to dissolve cells stained with MTT, and OD values were measured using a microplate reader at the wavelength of 570 nm. Relative cell viability was calculated according to the formula: OD value of experimental group/OD value of control group × 100%.
ELISA assay
Levels of TG, alanine aminotransferase (ALT), aspartate aminotransferase (AST), and gamma-glutamyl transpeptidase (GGT) were measured using commercial kits according to the manufacturers' instructions. ELISA kits for TG (SBJ-H0240), AST(SBJ-H0659), and GGT (SBJ-H0669) were obtained from Nanjing Senbeijia Biotechnology (Nanjing, China). An ELISA kit for ALT (E-EL-H0312c) was purchased from Elab science (Wuhan, China).
Western blot analysis
Total protein was extracted after cells were treated in the presence of OA and UDCA.Then, 30 μg of protein was used to perform immunoblotting. Primary antibodies against mSREBP-1 (AF6283), p-CRTC2 (AF8328), p-AKT (AF0016), and p-mTOR(AF3308) were purchased from Affinity Biosciences (OH, United States). Relative quantitative analysis of protein bands was performed using Quantity One analysis software (Bio-Rad).
Statistical analysis
Statistical analyses were performed using SPSS 21.0 (Chicago, IL, United States)software. All results were from three independent experiments, and are shown as the mean ± standard error of the mean. The results were analyzed with GraphPad Prism 5.0 (GraphPad Software Inc, United States) using one-way ANOVA.P<0.05 was regarded as significantly different.
RESULTS
UDCA administration improves OA-induced growth inhibition of LO2 cells
To determine the effect of UDCA on NAFLD, we first established a cell model of NAFLD. The oil red O staining analysis showed that 20 μg/mL OA significantly promoted the production of lipid droplets in LO2 cells, which suggested that we successfully developed the NAFLD cell model (Figure 1A, the right image). Given that OA treatment inhibited cell growth ability, we further evaluated the proliferation ability of the OA-induced LO2 cells treated with different concentrations of UDCA using MTT assay. When exposed to 20 μg/mL OA for 48 h, LO2 cell proliferation was inhibited significantly. This inhibition of proliferation by OA can be overcome to varying degrees by the addition of different levels of UDCA, especially under the condition of 2 mmol/L UDCA (cP<0.001, Figure 1B). Additionally, relative cell viability of LO2 cells prominently declined in the presence of OA at 24 h, 48 h, and 72 h in comparison with the control cells. However, when treated with 2 mmol/L UDCA for 24 h, 48 h, and 72 h, OA-mediated reduction of cell viability at different time points was notably upregulated; cell viability was close to the normal level at 72 h (bP< 0.01, Figure 1C). These data indicate that 2 mmol/L UDCA was effective for improving OA-induced cell growth inhibition.
UDCA treatment relieves OA-induced lipogenesis
The production of lipid droplets is closely related to various biochemical parameters.The levels of lipid metabolic indexes such as TG, ALT, AST, and GGT can accurately reflect the state of hepatic lipid metabolism. As shown in Figure 2A, OA treatmentinduced massive lipid accumulation of LO2 cells was dramatically suppressed under the treatment with UDCA for 72 h (Figure 2A, the middle imagevsthe right image).In addition, OA-induced LO2 cells showed a prominent increase of TG (1.5-fold), ALT(6-fold), AST (11-fold), and GGT (2-fold) levels compared to the control group. Upon the treatment with UDCA, the upregulation of ALT, AST, and GGT was significantly blocked (bP<0.01, Figure 2C-E), whereas UDCA treatment had no significant inhibitory effect on TG level in OA-exposed cells (Figure 2B). Although UDCA administration partly attenuated hepatic steatosis, there was a high level of lipid profile (TG, ALT, AST, and GGT) compared with the control group, which implied that adjuvant therapy was needed to better improve the disorder of hepatic lipid metabolism.
UDCA inhibits the expression of nSREBP-1 in the OA-induced NAFLD cell model
In the course of hepatic lipid metabolism, the cyclic adenosine monophosphate(cAMP) response elementbinding protein (CREB)regulated transcription coactivator 2(CRTC2) mediates the activation of mature SREBP-1 (nSREBP-1) which subsequently upregulates lipogenic gene expression and controls the biosynthesis of fatty acids and TG in hepatocytes[19]. To further confirm the protective effect of UDCA on excessive lipid synthesis, we determined the expression pattern of nSREBP-1 and activated CTRC2 in OA-exposed LO2 cells. Once treated with OA for 72 h, phosphorylated CRTC2 and mature SREBP-1 (nSREBP-1) showed a 1.5-fold and 2.5-fold increase,respectively (bP<0.01, Figure 3A and B, the second bandvsthe first band, the second boxvsthe first box, and the fifth boxvsthe fourth box). When exposed to UDCA, the enhancement of activated CRTC2 and SREBP-1 robustly declined compared with OA-exposed cells (cP<0.01, Figure 3A and B, the second bandvsthe third band, the second boxvsthe third box, and the fifth boxvsthe sixth box). These results suggest that CRTC2/SREBP-1 signaling transduction is implicated in the UDCA-mediated protective effect on lipid accumulation.
AKT/mTOR signaling pathway participates in the progression of UDCA-mediated improvement of lipid synthesis
It is known that AKT activation increases the activity of its downstream effector mTOR, which then promotes the activation of mature SREBP-1 by integrating
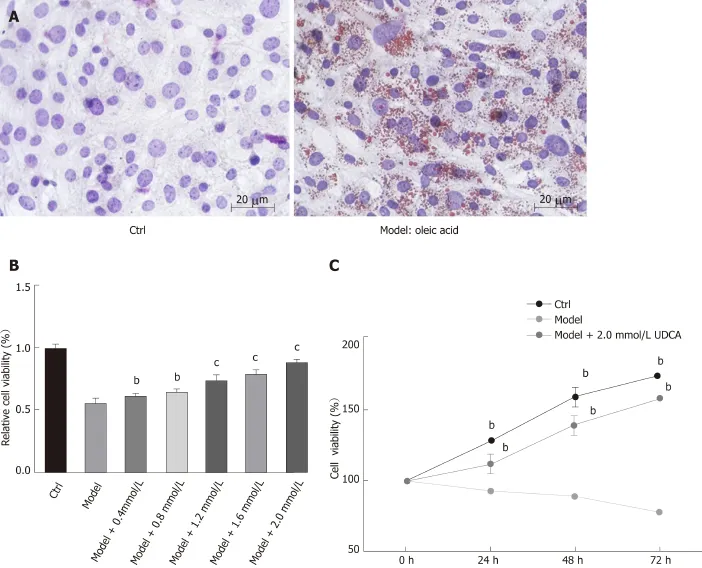
Figure 1 Effect of ursodeoxycholic acid on cell growth ability under the treatment of oleic acid. A: Oil red O staining of LO2 cells after oleic acid (OA) treatment for 72 h. B: Cell viability of OA-induced LO2 cells measured using the MTT assay under the condition of concentration gradient of ursodeoxycholic acid (UDCA) for 48 h. C: Cell viability of LO2 cells measured using the MTT assay under the condition of oleic acid and 2 mmol/L UDCA for 24 h, 48 h, and 72 h. Scar bar: 20 μm. Data are shown as the mean ± SE. bP<0.01, cP<0.001 vs model group. Ctrl: Control; UDCA: Ursodeoxycholic acid; OA: Oleic acid.
CRTC2[20,21]. Thus, we performed an immunoblotting assay to detect the changes of AKT/mTOR signaling and found that the activation of AKT and mTOR presented a 3-fold and 1.5-fold increase, respectively, under the condition of OA treatment for 72 h (Figure 4A and B, the second bandvsthe first band, the second boxvsthe first box,the fifth boxvsthe fourth box, and the eighth boxvsthe seventh box). UDCA administration recovered the levels of phosphorylated AKT and mTOR in OA-induced LO2 cells (aP<0.05,bP<0.01, Figure 4A and B, the second bandvsthe third band, the second boxvsthe third box, the fifth boxvsthe sixth box, and the eighth boxvsthe ninth box). Based on these results, we believe that UDCA ameliorates lipid metabolic disorder partly by regulating the AKT/mTOR signaling pathway.
DISCUSSION
There is increasing evidence that disorder of hepatic lipid metabolism is not only a hallmark of NAFLD, but also a biologic marker of cancer cells[22,23]. Numerous key enzymes involved in the progression ofde novofatty acid synthesis are enhanced and correlated with poor clinical outcomes in hepatic carcinoma (HCC)[24]. That means the importance of homeostasis of hepatic lipid metabolism is imaginable in liver diseases.In the present study, we found that the hydrophilic BA, UDCA, prevented lipogenesis by regulating the expression of mature SREBP-1 in LO2 cells, which suggested that UDCA administration might improve lipid metabolic disorder in NAFLD patients and slow the progression of metabolic disturbance-derived liver cancer.
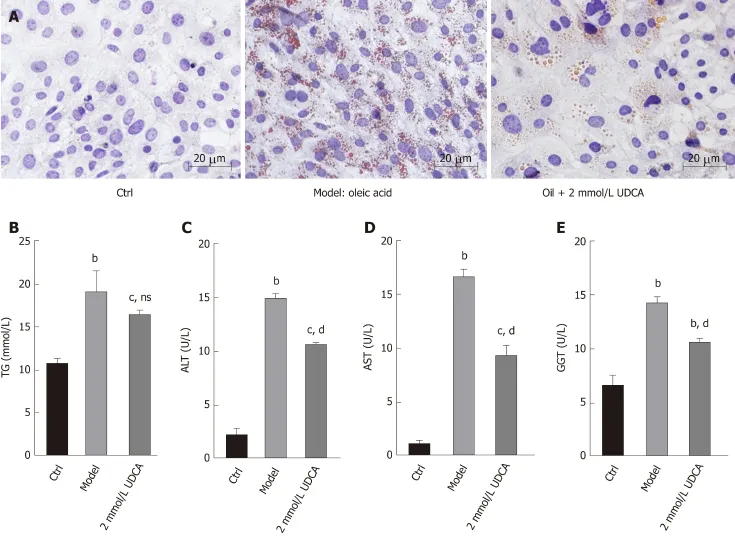
Figure 2 Impact of ursodeoxycholic acid on oleic acid-induced production of lipid profile. A: Oil red O staining of oleic acid-induced LO2 cells after ursodeoxycholic acid treatment for 72 h. B-E: Measurement of intracellular levels of triglycerides (B), alanine aminotransferase (C), aspartate aminotransferase (D),and gamma-glutamyl transpeptidase (E) by ELISA assay. Scar bar: 20 μm. Data are shown as the mean ± SE. bP<0.01, cP<0.001 vs ctrl group; ns vs model group.dP<0.001 vs model group. Ctrl: Control; UDCA: Ursodeoxycholic acid; OA: Oleic acid; TG: Triglycerides; ALT: Alanine aminotransferase; AST: Aspartate aminotransferase; GGT: Gamma-glutamyl transpeptidase; ns: No significant statistical difference.
Previous studies have pointed that OA-induced hepatic steatosis is always accompanied by oxide stress, cell apoptosis, and cell viability reduction in hepatocytes[25,26]. Oxidative stress acting as an apoptotic mediator of liver cells can cause damage of membrane lipid peroxidation and cell degeneration in the progression of NAFLD[27,28]. Here, we observed that 2 mmol/L UDCA significantly relieved the growth inhibition of OA-exposed LO2 cells, implying that 2 mmol/L was a safe and effective dosage forin vitrostudy. UDCA exerts a protective effect on NAFLD in rats by reducing the lipid profile including total cholesterol (TC) and TG[29].Treatment with UDCA suppressed activated liver X receptor α (LXRα)-mediated hepatic lipogenesis in high-fat diet (HFD)-fed mice[30]. These reports indicate that UDCA can serve as a regulator of lipid metabolic indexes in abnormal hepatic lipid metabolism-related liver diseases. Our findings indicated that administration with UDCA ameliorated OA-induced disturbance of liver dysfunction such as the increase of ALT, AST, and GGT levels in LO2 cells. UDCA treatment cannot result in a significant reduction of TG level in OA-induced cells, but had a certain regulatory role. Therefore, our data further supported the protective effect of UDCA in improving hepatocyte fatty degeneration and relieving the inflammatory reaction.
SREBP-1, a member of basic helix-loop-helix-leucine zipper transcription factor family, is mainly responsible for regulating fatty acid and cholesterol synthesis[31,32].SREBP-1 is synthesized as a precursor located on the nuclear membrane and endoplasmic reticulum[33]. Upon stimulus or sterol depletion, the precursor (fulllength SREBP-1) is cleaved into mature SREBP-1 which subsequently translocates to the nucleus (nSREBP-1) and activates transcription by binding to the promoter region of genes containing sterol regulatory element-1 (SREBP-1)[34,35]. Once SREBP-1 is activated, nSREBP-1 enhances the expression of cholesterol and fatty acid synthesis related genes[36]. In the course of lipid metabolism, activated AKT subsequently induces the activation of mTOR, which leads to the upregulation of its downstream CRTC2[37,38]. mTOR is an essential regulator of lipogenic metabolism by activating SREBP-1 cleavage[39]. CRTC2 as a mediator of the mTOR molecule, can modulate hepatic lipid metabolism by affecting COPII-dependent SREBP1 processing[21], which indicates that AKT-induced activation of mTOR can recruit CRTC2 as a complex to promote nSREBP-1 activity and the following lipogenesis. The present research demonstrated that CRTC2 activation, the AKT/MTOR signaling pathway, and nSREBP-1 expression were enhanced in the presence of OA. UDCA stimulation notably mitigated OA-initiated lipid metabolic disorder by suppressing this signaling pathway transduction. It is possible that UDCA attenuates OA-induced hepatic steatosis mainly by regulation of the AKT/mTOR/SREBP-1 signaling transduction.
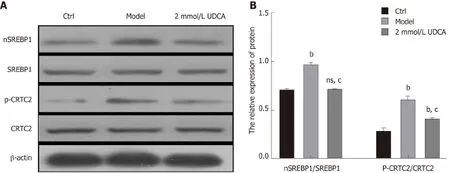
Figure 3 Alteration of CRTC2/SREBP-1 signaling pathway under the condition of ursodeoxycholic acid in oleic acid-induced LO2 cells. A: CRTC2 activity and nSREBP-1 expression detected by Western blot assay. B: Relative quantitative analysis of phosphorylated CRTC2 and mature SREBP-1 in LO2 cells. Data are shown as the mean ± SE. bP<0.01, ns vs ctrl group; cP<0.01 vs model group. Ctrl: Control; UDCA: Ursodeoxycholic acid; ns: no significant statistical difference.
In conclusion, we illustrated the protective role of UDCA in NAFLD. At a proper concentration of UDCA, OA-evoked lipid accumulation was nearly blocked by affecting SREBP-1 cleavage, implying that UDCA may contribute to developing more effective BA-based therapies for NAFLD. At present, the effects of UDCA on NAFLD in the clinic have seldom been reported, and especially the research of UDCA on liver histology is also limited. We carried out research at the cellular level to explore the AKT/mTOR/SREBP-1 signaling pathway as a new target for NAFLD treatment. Our research is expected to enrich the academic research content in this field and provide scientific basis for clinical application.
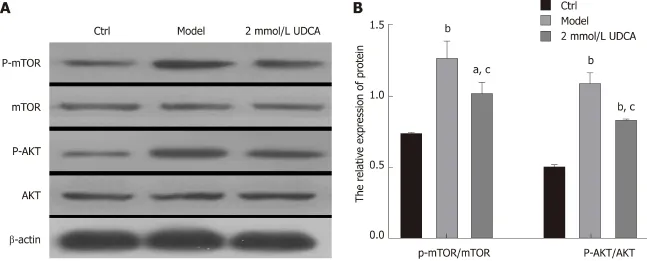
Figure 4 Effect of ursodeoxycholic acid on oleic acid-induced activation of the AKT/mTOR signaling pathway. A: Activation of AKT and mTOR detected by Western blot assay. B: Relative quantitative analysis of phosphorylated AKT and mTOR in LO2 cells. Data are shown as the mean ± SE. aP<0.05, bP<0.01 vs ctrl group; cP<0.01 vs model group. Ctrl: Control; UDCA: Ursodeoxycholic acid.

ARTICLE HIGHLIGHTS metabolism. This study has some limitations and further studies using animals and furtherin vitroresearch with agonists and inhibitors of mTOR as tools should be performed.
杂志排行

World Journal of Gastroenterology的其它文章
- International consensus statement on robotic hepatectomy surgery in 2018
- Growing burden of alcoholic liver disease in China: A review
- ?Esophageal diverticulum: New perspectives in the era of minimally invasive endoscopic treatment
- Gut microbiota profile in healthy Indonesians
- Risk factors for local recurrence and appropriate surveillance interval after endoscopic resection
- Accuracy of multi-echo Dixon sequence in quantification of hepatic steatosis in Chinese children and adolescents