以吡啶-2,5-二羧酸或噻吩-2,5-二羧酸为配体的过渡金属配合物的合成、晶体结构及性质
2019-04-12韩佳星梁楚欣李庆伟张淑芬
韩佳星 刘 峥*, 梁楚欣 唐 群*, 李庆伟 张淑芬,2
(1桂林理工大学化学与生物工程学院,电磁化学功能物质广西区重点实验室,桂林 541004)(2大连理工大学精细化工重点实验室,大连 116024)
近年来,过渡金属配合物在催化、吸附、药物、储能、材料等领域具有广泛的应用前景,利用不同的金属离子或者不同结构的有机配体,设计合成具有特定结构的功能配合物意义重大[1-8]。含杂环的羧酸配体在构筑过渡金属配合物时具有以下特点[9-14]:(1)由于配位点比羧酸化合物或杂环化合物多,会产生更多的配体模式;(2)调控羧基在杂环上的位置,可以选择对称的配体或不对称配体,形成的过渡金属配合物结构更加新颖;(3)增加杂环上羧基数量,可以构筑多核过渡金属配合物。另外,因为具有能与过渡金属离子形成螯合或双齿桥联的2个N原子,菲咯啉与4,4′-联吡啶被作为辅助配体广泛应用于配合物的结构构筑[15-16]。过渡金属配合物的合成方法有扩散法、水热法、溶剂挥发法、微波合成法等。其中水热法为反应物提供了一个高温高压的反应环境,能促进常温常压下难溶的有机配体和无机金属盐溶解,有利于合成结构新颖的配合物。而溶剂热法有利于配合物晶体的生长,且实验重复性强[17-18]。
本文选取吡啶-2,5-二羧酸和噻吩-2,5-二羧酸为主配体,菲咯啉和4,4′-联吡啶为辅助配体,与过渡金属盐通过水热法,设计合成了3种金属配合物。并通过单晶X射线衍射、红外光谱等测试方法对其结构进行了表征,同时研究了3种金属配合物的热稳定性和荧光性质。
1 实验部分
1.1 仪器与试剂
美国Perkin-Elmer 2400CHNS/O元素分析仪;德国耐驰公司TG-DSC热分析仪;日本岛津公司ShimadzuFTIR-8400红外光谱仪;荷兰帕纳科(PANalytical)公司X射线单晶衍射仪;日本日立公司F-4600荧光光谱仪。吡啶-2,5-二羧酸、噻吩-2,5-二羧酸、Cu(NO3)3·3H2O、Co(NO3)2·6H2O、Ni(CH3COOH)2·4H2O、4,4′-联吡啶、菲咯啉均为分析纯。
1.2 配合物的合成
1.2.1 [Co2(L1)2(bipy)(H2O)6]·bipy·H2O(1)的合成
将 15 mL 含有 0.5 mmol(0.145 6 g)的 Co(NO3)2·6H2O、0.5 mmol(0.083 56 g)的吡啶-2,5-二羧酸(2.5-bipy,H2L1)、0.5 mmol(0.078 1 g)的 4,4′-联吡啶(4,4′-bipy)和0.5 mmol(0.02 g)NaOH的水溶液在室温下磁力搅拌1 h,然后将烧杯中的混合液转移至25 mL带有聚四氟乙烯衬底的水热反应釜中[19]。将反应釜放入烘箱中,加热至140℃,然后晶化72 h。72 h后进行程序降温,以10℃·h-1的速度降温到100℃,再保温10 h,自然冷却至室温。有紫红色块状晶体生成,过滤,室温下干燥。产率72%(基于Co)。元素分析实测值(括号内为按C34H38N6Co2O16计算值,%):C 45.26(45.14),H 4.19(4.24),N 9.31(9.24)。 IR(KBr,cm-1):3 450(-OH),1 600(C=O),1 300(COO-吸收峰),1 250~1 150(杂环的伸缩振动峰),1 240(C=N),700(Co-O),600(Co-N)。
1.2.2 [Cu(L2)2(bipy)2]n(2)的合成
将 15 mL 含有 0.5 mmol(0.072 5 g)的 Cu(NO3)3·3H2O、0.5 mmol(0.086 1 g)的噻吩-2,5-二羧酸(2,5-tdc,H2L2)和 0.5 mmol(0.0781 g)的 4,4′-联吡啶(4,4′-bipy)的水溶液混合均匀,再往其中加入4 mL CH3OH,在室温下磁力搅拌1 h。然后将烧杯中的混合液转移至25 mL带有聚四氟乙烯衬底的水热反应釜中。将反应釜放入烘箱中,加热至140℃,然后晶化72 h。72 h后进行程序降温,以10℃·h-1的速度降温到100℃,再保温10 h,自然冷却至室温。有蓝色针状晶体生成,过滤,室温下干燥。产率72%(基于Cu)。元素分析实测值(括号内为按C16H10CuN2O4S计算值,%):C 49.27(49.30),H 2.56(2.59),N 7.21(7.18),S 8.19(8.23)。 IR(KBr,cm-1):3 400(-OH),3 150(芳环 C-H 伸缩振动),1 650(C=O),1 550(芳环 C=C),1 350(C=N),880、700(芳环 C-H 面外弯曲),750(Cu-N),500(Cu-O)。
1.2.3 [Ni2(L2)(phen)2(H2O)4](3)的合成
将6 mL含有1 mmol(0.249 g)的 Ni(CH3COOH)2·4H2O、0.5 mmol(0.086 1 g)的噻吩-2,5-二羧酸(2,5-tdc,H2L2)和 0.5 mmol(0.1 g)的菲咯啉(1,10-phen)的水溶液混合均匀,再往其中加入4 mLCH3OH,在室温下磁力搅拌30 min。用氨水调节pH值至5~6之间,再继续搅拌30 min。然后将烧杯中的混合液转移至25 mL带有聚四氟乙烯衬底的水热反应釜中,将反应釜放入烘箱中,加热至140℃然后晶化72 h。72 h后进行程序降温,以10℃·h-1的速度降温到100℃,再保温10 h,自然冷却至室温。有亮绿色针状晶体生成,过滤,室温下干燥。产率72%。元素分析实测值(括号内为按C36H20N4Ni2O12S2计算值,%):C 48.51(48.53),H 3.15(3.14),N 6.32(6.29),S 7.15(7.18)。 IR(KBr,cm-1):3 400(-OH),1 680(C=O),1 460(芳环 C=C),1 300(C=N),800、700(芳环 C-H 面外弯曲),900(Ni-O),600(Ni-O)。
1.3 晶体结构测试
选取尺寸分别为0.21 mm×0.16 mm×0.11 mm(1)、0.33 mm×0.28 mm×0.13 mm(2)和 0.22 mm×0.16 mm×0.31 mm(3)的晶体,用 Bruker SMART APEX CCD单晶衍射仪,采用经石墨单色化的Mo Kα射线(λ=0.071 073 nm),用 φ-ω 扫描模式收集配合物的衍射点。衍射的强度数据用SADABS程序[20-21]进行经验吸收校正。晶体的结构用SHELXS-97[22]程序通过直接法解析出来,对全部的非氢原子的坐标及其各向异性参数用SHELXS-97程序进行全矩阵最小二乘法修正。所有的氢原子都为理论上加氢。有关晶体学数据见表1。
CCDC:1508171,1;1508172,2;1508114,3。
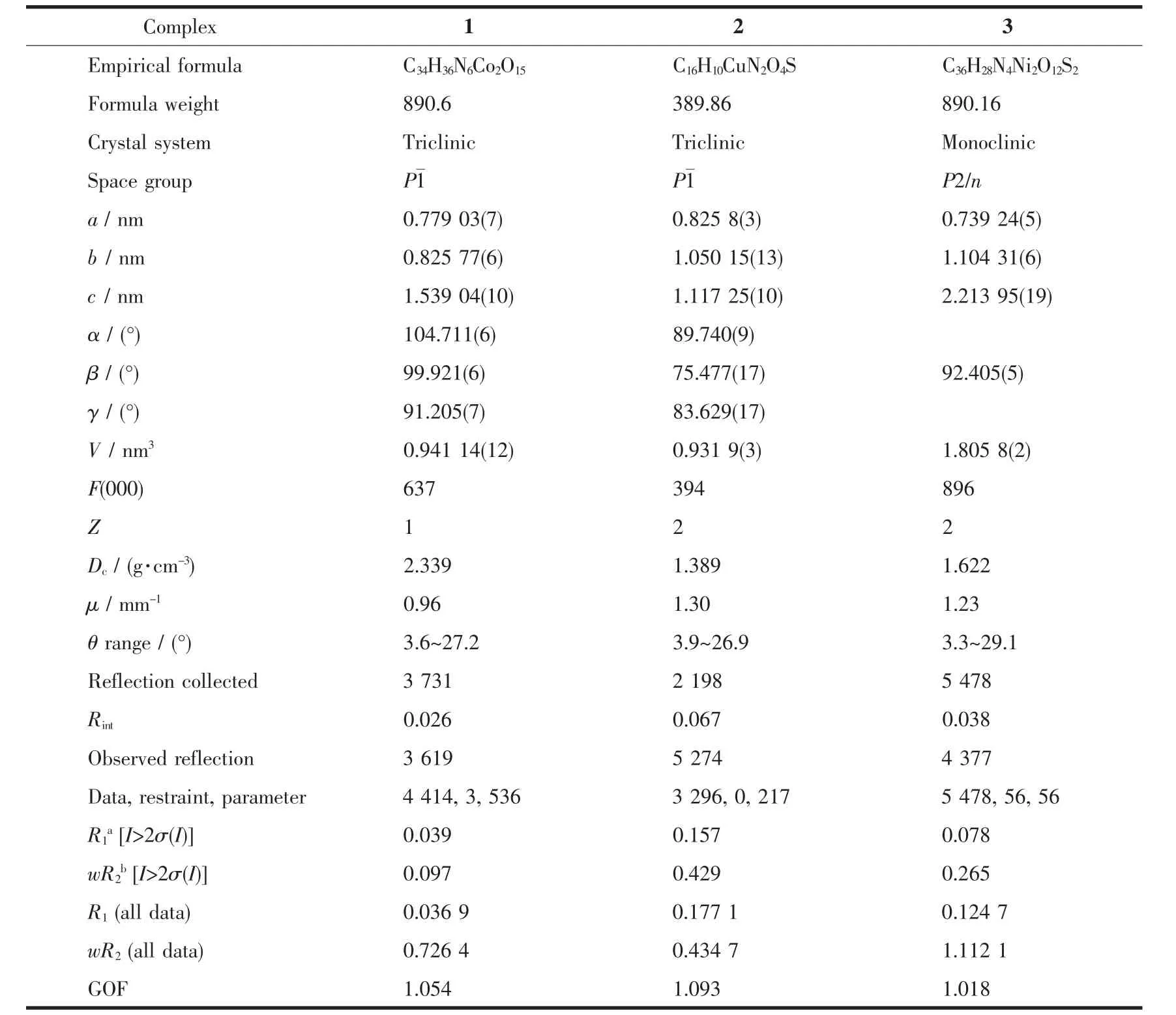
表1 配合物1~3的晶体学数据Table 1 Crystallographic data of complexes 1~3
2 结果与讨论
2.1 晶体结构描述
2.1.1 配合物1的晶体结构
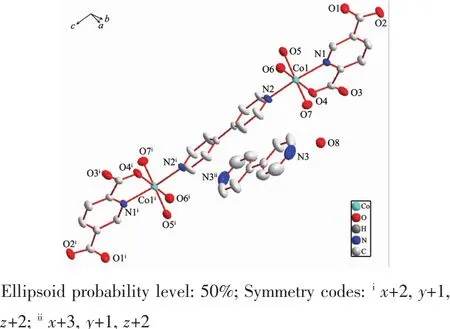
图1 配合物1的分子结构Fig.1 View of molecular structure of complex 1
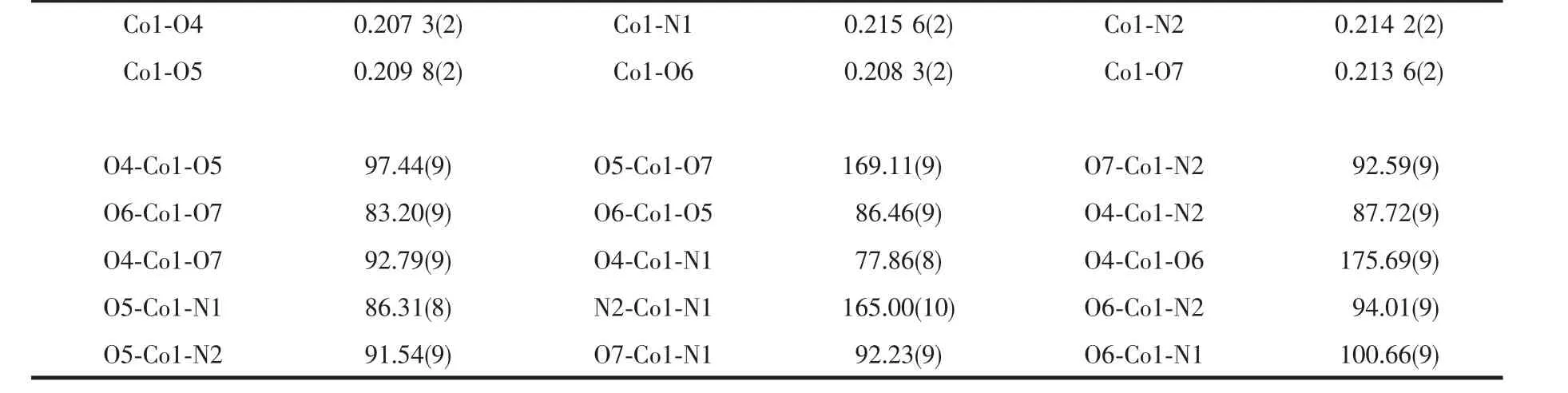
表2 配合物1的主要键长(nm)和键角(°)Table 2 Selected bond lengths(nm)and bond angles(°)of complex 1
配合物1的分子结构如图1所示,主要键长和键角见表2。配合物1属于三斜晶系,P1空间群。由图1可知,配合物1结构单元由2个中心离子(Co2+)、2 个吡啶-2,5-二羧酸阴离子 (L12-)、2 个 4,4′-联吡啶中性分子和7个水分子组成。配合物1的钴离子配位环境如图2,吡啶-2,5-二羧酸根阴离子通过吡啶环上的氮原子与邻近羧基氧原子以双齿螯合的方式与钴离子配位。其中一个4,4′-联吡啶中性分子以双齿桥联的方式连接2个钴离子。另一个4,4′-联吡啶则以游离的方式存在。钴离子分别与吡啶-2,5-二羧酸的1个氮原子(N1)和1个羧基氧原子(O2),与 4,4′-联吡啶的 1 个氮原子(N2)以及 3 个水分子(O5、O6、O7)发生配位。6个配位原子在钴离子周围构成了畸变八面体构型。其中,N2、O2、N3、O5构成赤道平面,O6、O7呈轴向分布。该配合物通过分子间弱的范德华力、氢键等作用力构成了三维空间构型。图3为配合物1的a轴方向堆积图。
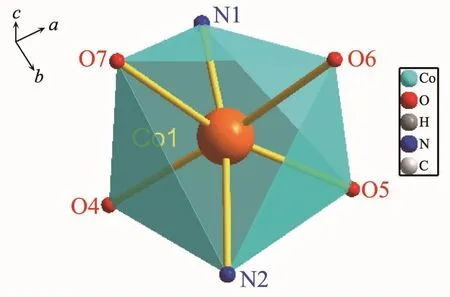
图2 配合物1的Coギ配位多面体图Fig.2 Coordination polyhedron for Coギof complex 1
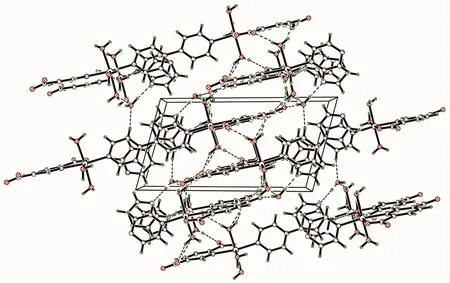
图3 配合物1的a轴方向的堆积图Fig.3 Packing diagram of complex 1 viewed along a axis
2.1.2 配合物2的结构分析
配合物2的分子结构图见图4,主要键长和键角见表3。配合物2属于三斜晶系,P1空间群。由图4可知,配合物2结构单元由1个中心离子(Cu2+)、2个噻吩-2,5-二羧酸阴离子(L22-)、2 个 4,4′-联吡啶中性分子组成。配合物2的铜离子配位环境如图5所示,在中心铜离子的配位环境中,O1、O2、O4i分别来自2个不同的噻吩-2,5-二羧酸中的羧基氧原子,同时 N1i、N2 来自辅助配体 2 个不同的 4,4′-联吡啶上的氮原子,形成三角双锥构型。通过氢键及π-π堆积的弱相互作用力构成了配合物2的三维网状空间结构。图6为配合物2在a方向上的堆积图。Symmetry codes:ix,y,z+1;iix+1,y+2,z+1;iiix,y-1,z;ivx,y,z-1;vx,y+1,z.
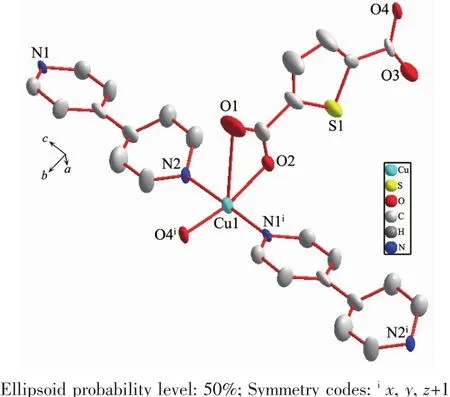
图4 配合物2的结构单元图Fig.4 Crystal structure unit of complex 2
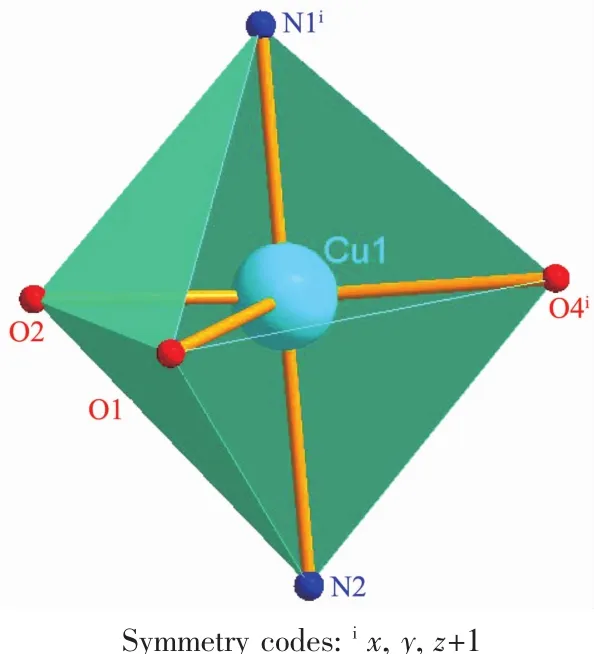
图5 配合物2的Cuギ配位多面体图Fig.5 Coordination polyhedron for Cuギof complex 2

表3 配合物2的主要键长(nm)和键角(°)Table 3 Selected bond lengths(nm)and bond angles(°)of complex 2
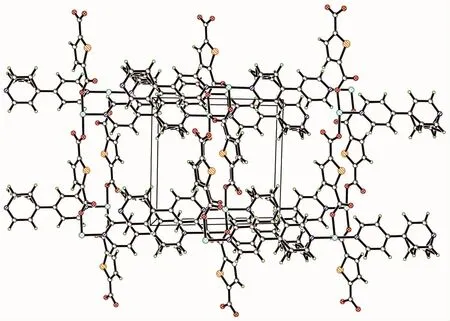
图6 配合物2的a方向的堆积图Fig.6 Packing diagram of complex 2 viewed along a axis
2.1.3 配合物3的结构分析
配合物3的分子结构如图7所示,主要键长和键角见表4。由图7可知,配合物3最小结构单元由2 个 Ni2+,2 个噻吩-2,5-二羧酸根阴离子(L22-),2 个菲咯啉中性分子,2个氢氧根离子和2个配位水分子组成。中心离子Ni1分别与1个噻吩-2,5-二羧酸根阴离子的羧基(O1)以单齿形式配位,与1个中性菲咯啉分子(N1、N2)以双齿螯合的形式配位,与2个氢氧根离子(O6,O6i)以单齿桥联的形式配位,以及与1个水分子单齿配位。6个配位原子在镍离子周围形成了畸变八面体构型(图8),其中N1、O5、O6和O6i占据赤道平面,O1和N2占据轴向位置。氢键及π-π堆积的弱相互作用力构筑了配合物3的三维网状空间结构。图9为配合物3在a方向上的堆积图。

图7 配合物3的结构单元图Fig.7 Crystal structure of complex 3
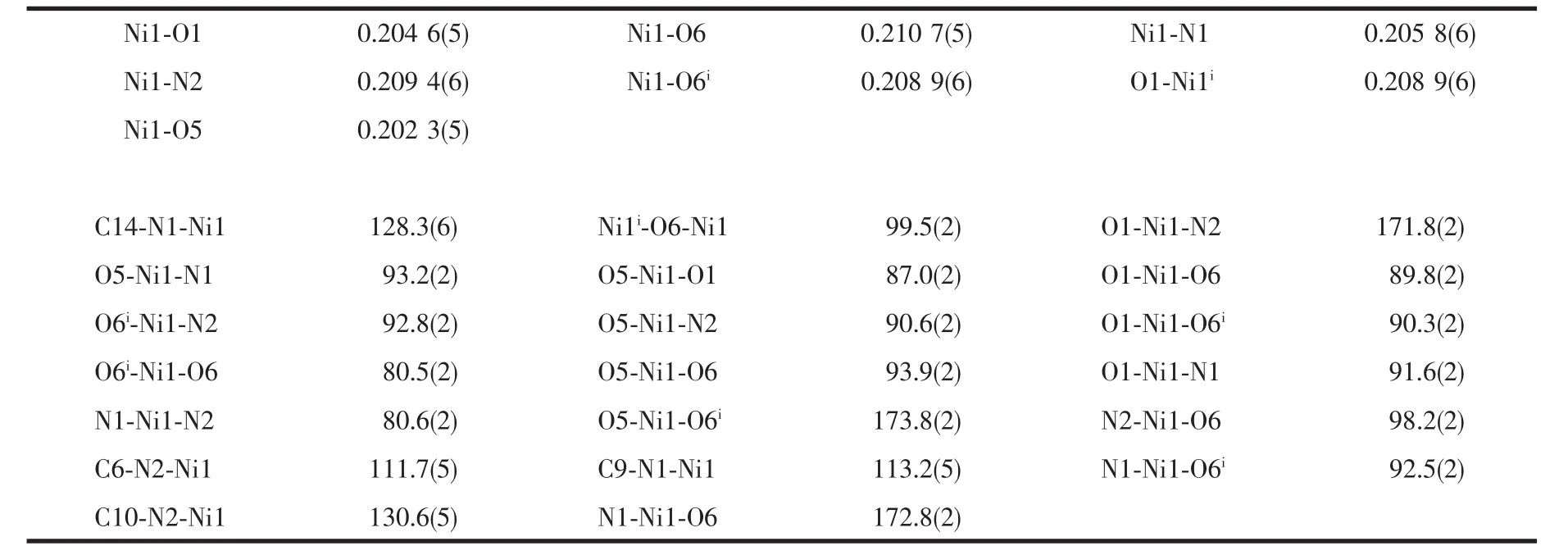
表4 配合物3的主要键长(nm)和键角(°)Table 4 Selected bond lengths(nm)and bond angles(°)of complex 3
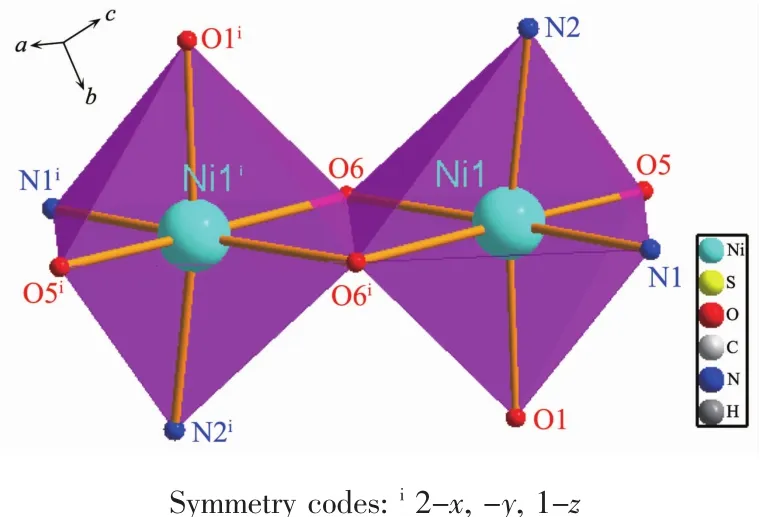
图8 配合物3的Niギ配位多面体图Fig.8 Coordination polyhedron for Niギof complex 3
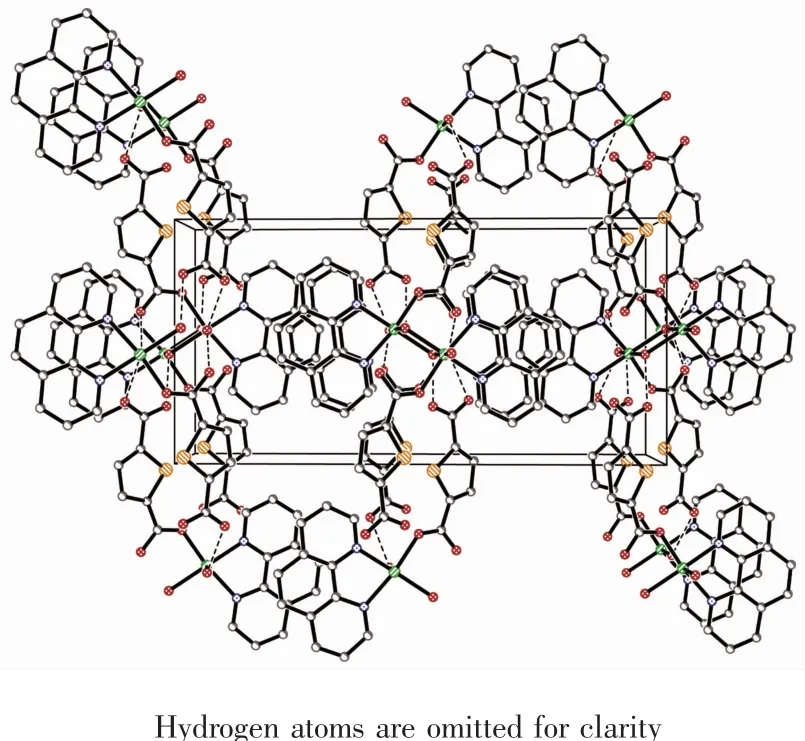
图9 配合物3的a方向的堆积图Fig.9 Packing diagram of complex 3 viewed along a axis
2.2 配合物材料的荧光性质研究
图10 ~12分别为配合物1(激发波长为376 nm),2(激发波长为402 nm)和3(激发波长为440 nm)及相应配体的荧光发射光谱。由图10~12可知,配合物1、配合物2和配合物3最大发射峰分别位于λEm=468、469和470 nm,与相应的配体相比,发射波长红移(配体吡啶-2,5-二羧酸、噻吩-2,5 二羧酸、菲咯啉、4,4′-联吡啶的最大发射峰分别位于 λEm=321、385、362和327 nm)。配体形成配合物后,谱带形状、强度有所增强,发射波长红移。这是由于配体与金属离子形成配合物结构,刚性和共平面程度增强,增大了共轭体系,因而引起发射波长红移,有较多的电子回到基态,引起荧光强度增强[23-24]。配合物2和3的最大发射峰波长非常相近,说明发光主要是基于主配体噻吩-2,5二羧酸本身的发光。
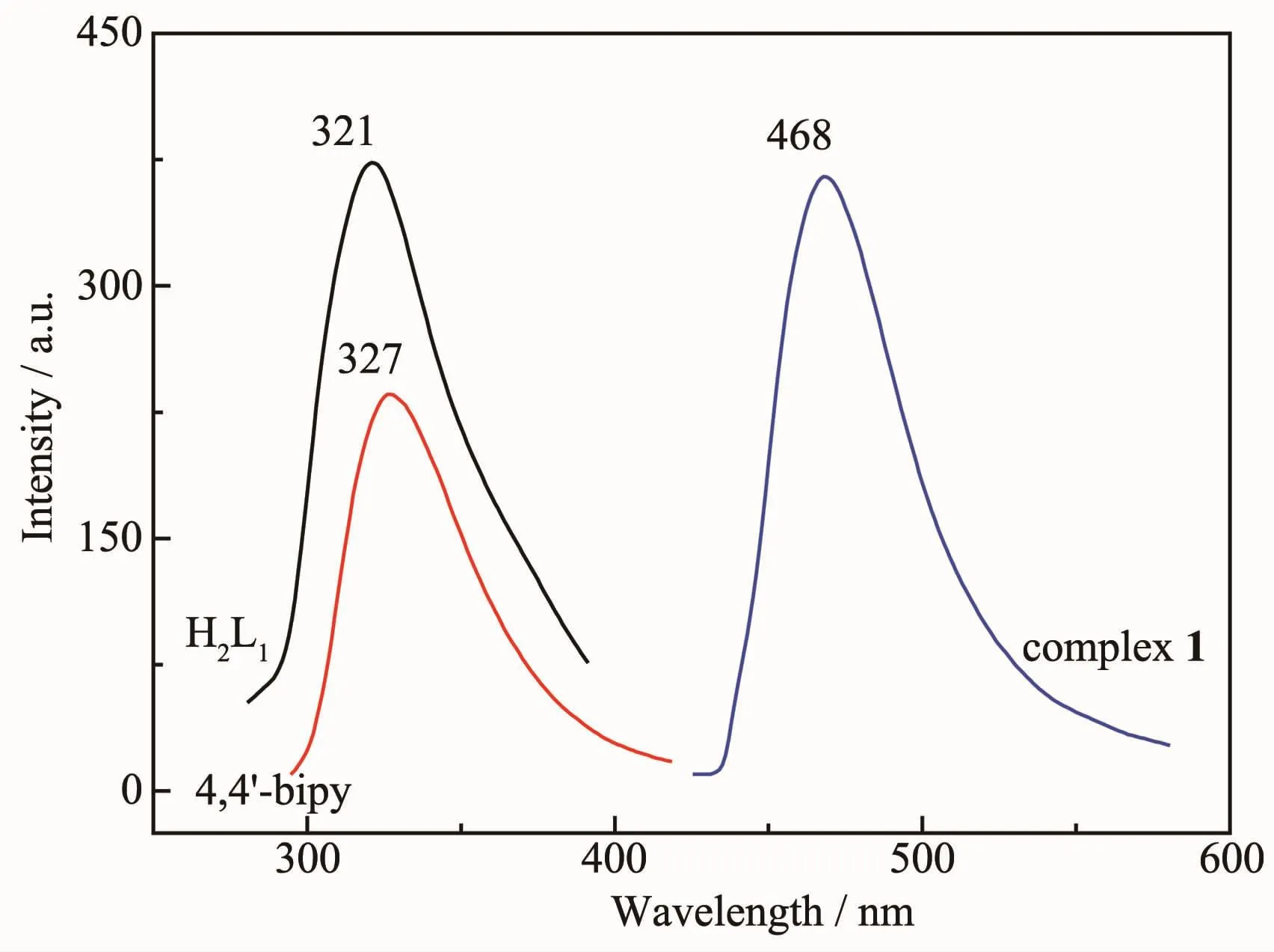
图10 配体H2L1、4,4′-联吡啶和配合物1的发射光谱Fig.10 Emission spectra of H2L1,4,4′-bipy and complex 1
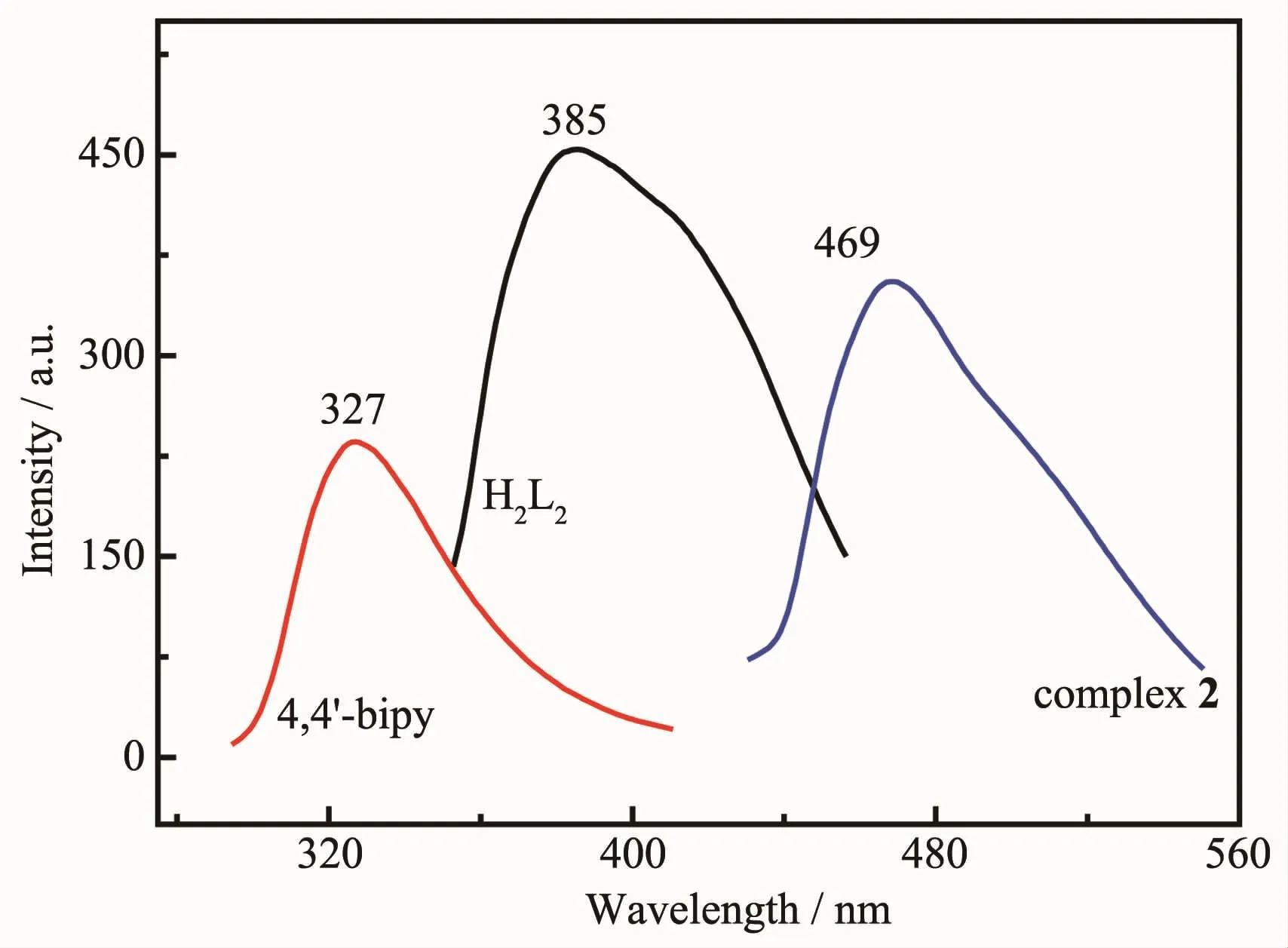
图11 配体H2L2、4,4′-联吡啶和配合物2的发射光谱Fig.11 Emission spectra of H2L2,4,4′-bipy and complex 2
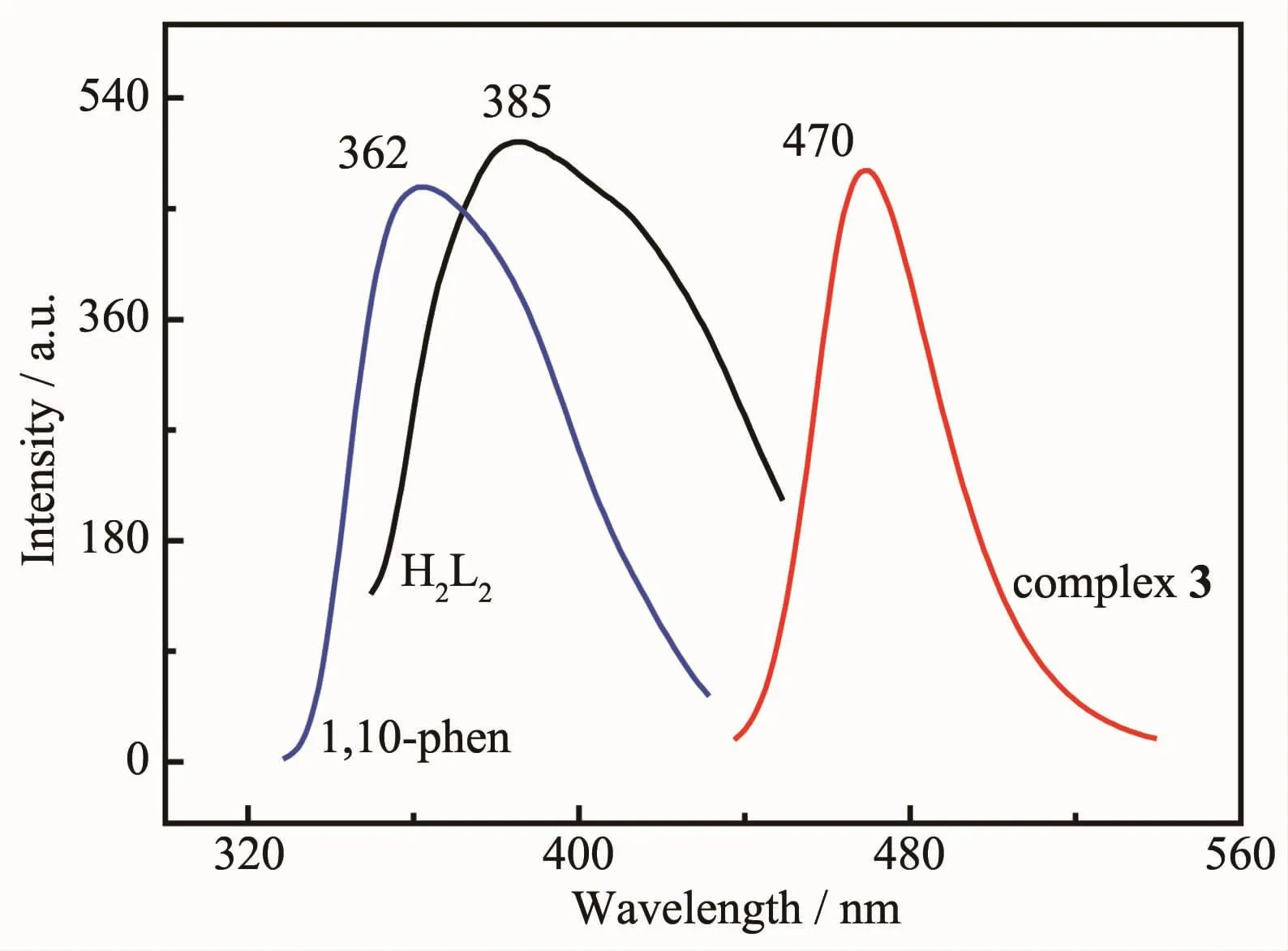
图12 配体H2L2、菲咯啉和配合物3的发射光谱Fig.12 Emission spectra for H2L2,1,10-phen and complex 3
2.3 配合物的热稳定性研究
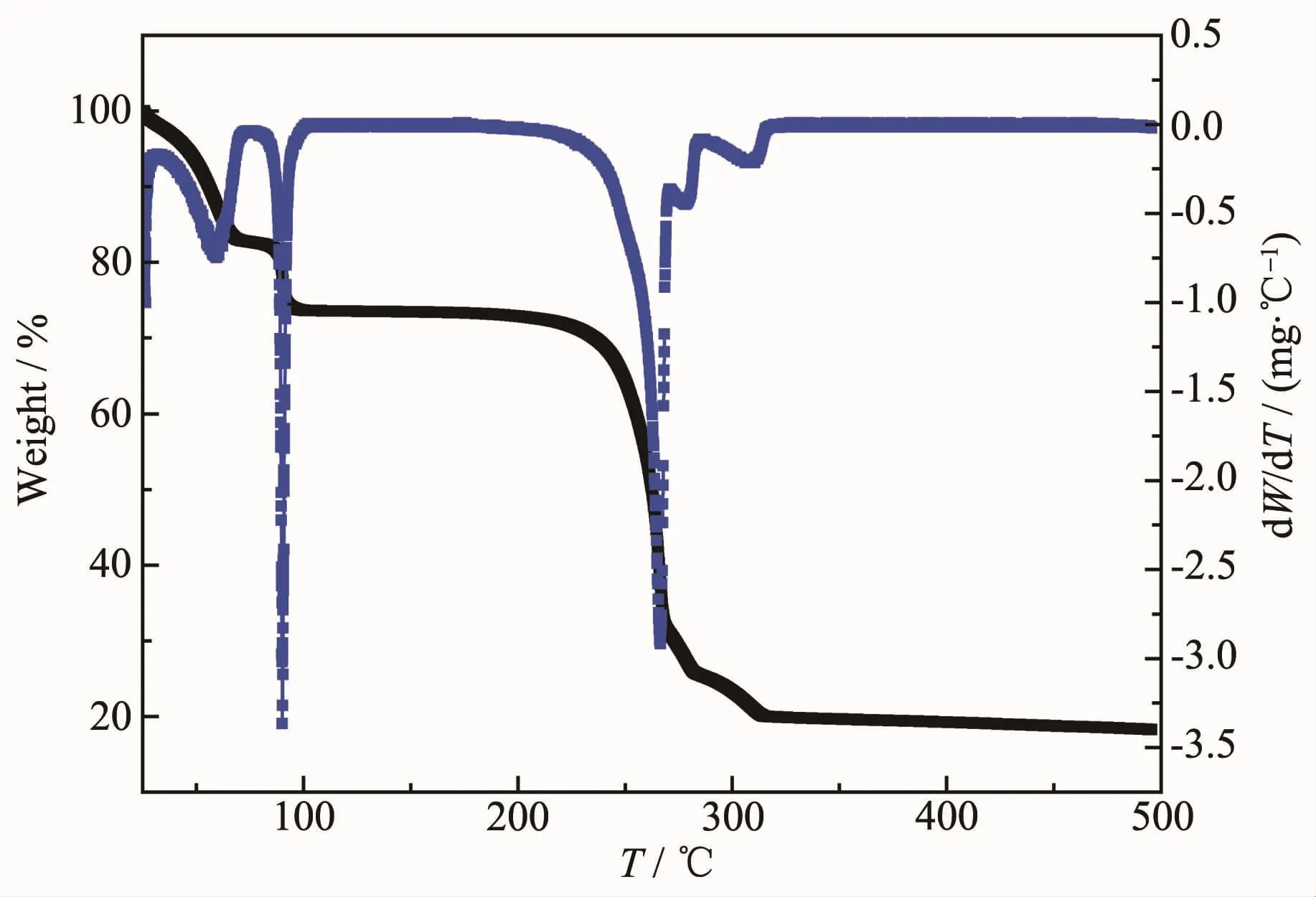
图13 配合物1的TG/DTG曲线Fig.13 Thermal gravimetric analysis(TG/DTG)for complex 1
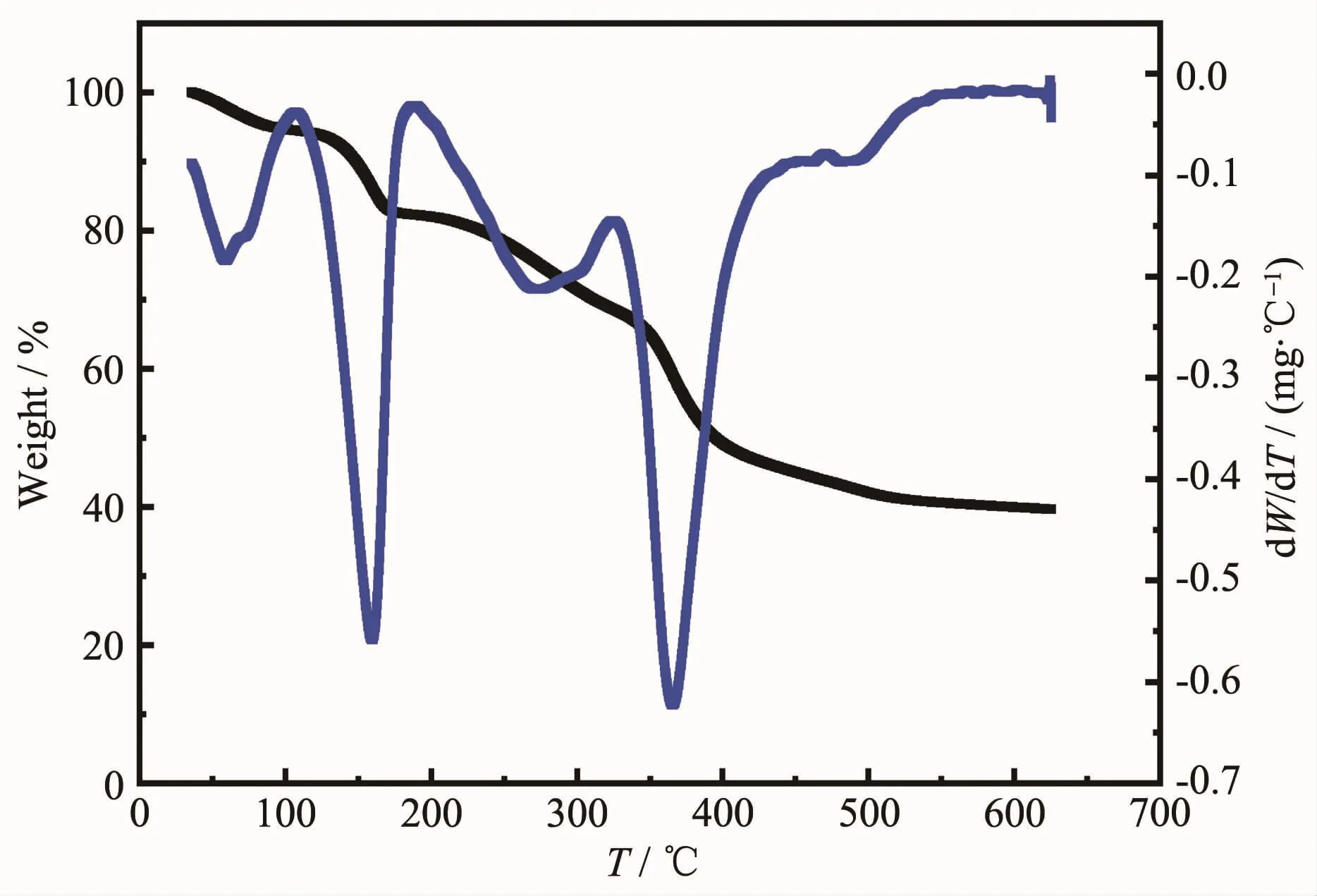
图14 配合物2的TG/DTG曲线Fig.14 Thermal gravimetric analysis(TG/DTG)for complex 2
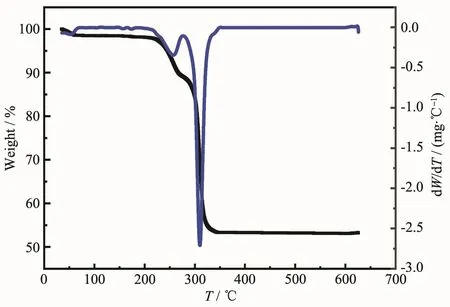
图15 配合物3的TG/DTG曲线Fig.15 Thermal gravimetric analysis(TG/DTG)for complex 3
三种配合物以及相应的配体的TG/DTG曲线如图13~15所示。由图13可知,配合物1的热重曲线表明,配合物1在室温至100℃时,失重率为26.25%,归属于配合物1晶体中的1个游离的4,4′-联吡啶和1个游离水分子的脱去,理论失重率为19.26%,实验测试失重率与理论计算失重率有一定的偏差,可能是样品中含有少量吸附水导致实验测试失重率偏高;在100~250℃范围内,TG曲线趋于平台,表明此时样品没有发生分解;随后TG曲线急速下降,表明样品的主体骨架开始分解;300℃后曲线不再下降,表明配合物1不再发生分解,最后的残余量为19.93%,剩余物可能为CoO(理论值:16.56%)。
由图14可知从室温到100℃区间,配合物2失重率为5.35%,归属于配合物2晶体吸附水的失去;随后配合物2的TG曲线逐渐下降,表明配合物2的骨架开始分解;550℃之后,TG曲线趋于平缓,不再下降,表明配合物2不再分解,残余率为39.61%,理论计算值为20.40%(CuO),残余率高于理论计算值,可能是由于配合物含碳量较高,产生积碳效应[25]。
由图15可知从室温到100℃区间,配合物3的失重率为1.53%,归属为配合物3吸附水的失去;在100~220℃区间,TG曲线出现平台,表明此时样品没有发生分解,随后TG曲线下降,表明配合物3骨架开始分解;350℃之后,曲线趋于平台,表明配合物3不再分解,残余率为53.24%,理论计算值为37.16%(Ni2O3),残余率高于理论计算值,可能是由于配合物含碳量较高,产生积碳效应[25]。
3 结 论
以 2 种主配体 2,5-bipy、2,5-tdc和 2 种辅助配体 1,10-phen、4,4′-bipy 与金属盐反应,成功合成出3种金属配合物材料。配合物1和配合物3均为零维双核小分子结构,配合物2为二维层状结构。用荧光光谱(FL)分析了3个配合物材料的荧光性质,结果表明配体形成配合物材料后,发射波长红移,荧光强度增强。
