西北高海拔地区放养偶蹄类动物肠道微生物多样性的宏基因组比较研究
2019-04-11尚立强薛世魁王惜婧张小丽
尚立强,薛世魁,王惜婧,张小丽*
(1.甘肃农业大学动物医学院,甘肃兰州 730070;2.甘肃省兰州市城关区动物卫生监督所,甘肃兰州 730030)
宏基因组学(metagenomics)是一种以新一代高通量测序技术为基础,以特定环境下微生物群体基因组为研究对象,分析微生物群体的多样性、种群结构、进化关系、功能活性、相互协作关系及其与环境的关系,发掘潜在的生物学意义。与传统微生物研究方法相比,宏基因组测序技术规避了绝大部分微生物不能培养、痕量菌无法检测的缺点,可从基因层面对整个微生物群落进行分析,并可使序列结果分析的速度与准确性都大大提高[1]。目前,该技术已被成功应用于研究污水、植物、动物及人体等多种环境中微生物的多样性[2-5]。通过宏基因组测序分析技术不仅可以明确宿主与微生物之间的相互关系,提高宿主的健康状态和营养水平,而且可以增加对宿主胃肠道微生物代谢通路的了解。近年来,关于动物肠道微生态的研究受到人们广泛关注,研究动物肠道微生物不仅可以描述动物肠道微生物的系统发育,分析其肠道菌群功能,了解其胃肠道微生物区系的变化规律、信号通路及其响应机制,而且可以通过改变肠道微生物组成、增加优势菌群数量等有效措施预防和治疗某些肠道疾病,进而为动物健康、可持续的繁殖和生存做出贡献,同时也为研究动物生存环境的微生物多样性提供了相关数据[6-7]。
武威市地处我国西北地区,位于101°49′~104°16′E,36°29′~39°27′N,拥有南部祁连山山地、中部走廊平原和北部荒漠3个地貌单元,海拔介于1 020~4 874 m。双峰骆驼、绵羊及白牦牛是这一地区主要放养的经济动物。其中,天祝白牦牛是我国稀有而珍贵的地方牦牛类群,是经过长期自然选育和人工选育而成的特有畜种,已被列入国家级畜禽保护品种,其肠道微生物的多样性具有很强的地域代表性。通过研究这些动物肠道微生物的多样性,可为进一步了解我国西北高海拔地区特有的环境微生物多样性提供参考依据。笔者采集了这3种动物的粪便样品并提取其总DNA,通过宏基因组denovo测序和生物信息学分析,阐明并比较了这些动物肠道微生物的多样性,并对其群落功能进行了分析。
1 材料与方法
1.1主要试剂与仪器琼脂糖、PBS、抗生素、TaqMaster Mix、DNA Marker 2000,均购自上海生工生物工程技术有限公司;NanoDrop2000购自Thermo Scientific公司。
1.2粪便样品采集从甘肃省武威市采集新鲜的双峰骆驼、绵羊及白牦牛粪便样品各15份。样品采集时避开外部杂菌污染区对中部的粪便样品进行无菌采集后储存于冰盒中,立即送至实验室进行处理。
1.3DNA提取及检测利用QIAamp DNA Stool Mini Kit对样品总DNA进行提取,所有操作按照试剂盒提供的操作说明书进行。DNA提取完毕后,利用1%琼脂糖凝胶电泳对抽提的基因组DNA完整性进行了检测。同时,利用NanoDrop2000超微量分光光度计(Thermo Scientific,USA)和TBS-380微型荧光计(Promega,USA)对抽提DNA样品的纯度和浓度分别进行了测定。条带单一或稍有弥散,无色素、RNA、蛋白等杂质污染,总量满足2次建库需求的DNA样品方可进行后续测序分析。
1.4上机文库构建及测序检测合格的DNA样品由美吉生物公司进行文库构建及测序。利用Covaris M220超声发生器将合格的DNA样品打断成300 bp左右的片段,然后使用TruSeq Nano DNA Sample Preparation Kits选择性回收目的片段并在两端连接接头,以构建测序文库。文库建好后,使用Illumina Hiseq2500平台进行双末端测序,测序得到的下机数据(也称为原始数据RawData)用于后续的生物信息分析。
1.5序列处理及生物信息学分析为了提高后续分析质量和可靠性,首先对原始序列进行了拆分、质量剪切以及去除污染等优化处理。使用软件Seqprep(https://github.com/jstjohn/SeqPrep)去除含N碱基的reads,对序列3’端和5’端进行质量剪切,然后使用软件Sickle(https://github.com/najoshi/sickle)去除剪切后长度小于50 bp及平均质量值低于20的reads,保留高质量的pair-end reads和single-end reads,最后通过BWA软件(http://bio-bwa.sourceforge.net)将reads比对宿主DNA序列,去除与宿主基因组相似性高的污染reads。将优化处理后的序列使用拼接软件SOAPdenovo(http://soap.genomics.org.cn/,Version 1.06)进行拼接组装。拼接主要参数k-mer值设范围为39~47。在scaffolds内部gap处,将scaffolds打断成新的contigs,并对大于等于500 bp的contigs进行统计,从中选择最优组装结果。使用MetaGene(http://metagene.cb.k.u-tokyo.ac.jp/)对拼接结果中的contig进行基因预测。
然后,对获得的基因序列进行物种相对丰度和功能上的分类及注释。使用BLASTP程序(BLAST Version 2.2.28+,http://blast.ncbi.nlm.nih.gov/Blast.cgi)将基因集序列分别与eggNOG数据库(http://eggnog.embl.de/)和KEGG 数据库( http://www.genome.jp/kegg/)进行比对(e值为1e-5),进而获得详细的物种注释。此外,在上述分析的基础上,进行了相似聚类,分组排序、差异比较等多方向的统计分析和探索,并对结果进行了可视化展示,挖掘数据中的有效信息。
2 结果与分析
2.1门水平上不同动物肠道微生物的比较该研究对双峰骆驼、绵羊和白牦牛的粪便样品进行了宏基因组学分析,每个样本平均产生15 Gbp的测序数据。分类学分析结果显示,在门水平上双峰骆驼、绵羊和白牦牛的肠道微生物群落主要分布于以下7个优势菌门(占比大于1%),包括拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、纤维杆菌门(Fibrobacteres)、变形菌门(Proteobacteria)、螺旋体门(Spirochaetes)、疣微菌门(Verrucomicrobia)和广古菌门(Euryarchaeota)。其中Bacteroidetes在3种动物肠道中相对丰度都是最高的(骆驼64.61%,绵羊63.95%,白牦牛77.09%),其次是Firmicutes(骆驼21.53%,绵羊7.54%,白牦牛12.48%)。这表明草食动物在肠道微生物多样性上存在一致性。其余5个优势门类在3种动物中的相对丰度各不相同(图1a)。Fibrobacteres在骆驼、绵羊和白牦牛中的占比分别为3.41%、7.54%和0.08%;Proteobacteria在骆驼、绵羊和白牦牛中的占比分别为2.48%、4.71%和4.15%;Spirochaetes在骆驼、绵羊和白牦牛中的占比分别为0.68%、5.03%和0.68%;Verrucomicrobia在骆驼、绵羊和白牦牛中的占比分别为1.96%、1.41%和2.41%;Euryarchaeota在骆驼、绵羊和白牦牛中的占比分别为0.15%、1.60%和1.60%。
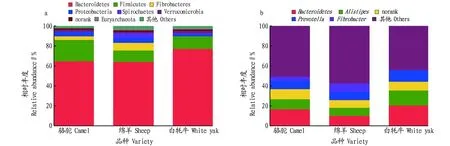
图1 门(a)和属(b)水平上不同动物肠道微生物的比较 Fig.1 Comparison of intestinal microbe among different animals at phylum(a) and genus(b) levels
2.2属水平上不同动物肠道微生物的比较在属水平上,3种动物的肠道微生物群落主要分布于以下4个优势属(占比大于10%),包括拟杆菌属(Bacaeroides)、Alistipes、普氏菌属(Prevotella)和 纤维杆菌属(Fibrobacter),其中Bacteroides在3种动物肠道中的相对丰度都是最高的(骆驼28.09%,绵羊22.62,白牦牛31.43%)(图1b)。利用韦恩图展示3种动物各自特有和共有细菌物种数目,结果显示这3种动物的肠道微生物种类在分类学水平属上存在高度的相似性(图2)。骆驼、绵羊和白牦牛肠道中所鉴定到的总的属数目分别为1 393、1 223和1 404个,而其中3种动物所共有的属数目多达1 067个,说明这一地区的环境微生物群落具有一定的稳定性。
2.3COG功能分析为了调查3种不同动物之间的肠道微生物基因功能差异,将基因集序列与eggNOG 数据库进行比对(e值为1e-5)获得基因对应的COG(Clusters of orthologous groupsof proteins),然后使用COG对应的基因丰度总和计算该COG的丰度。结果显示,3种动物都有20个 functional COG categories 被检测到,而且它们之间的丰度存在高度的相似性(图3)。
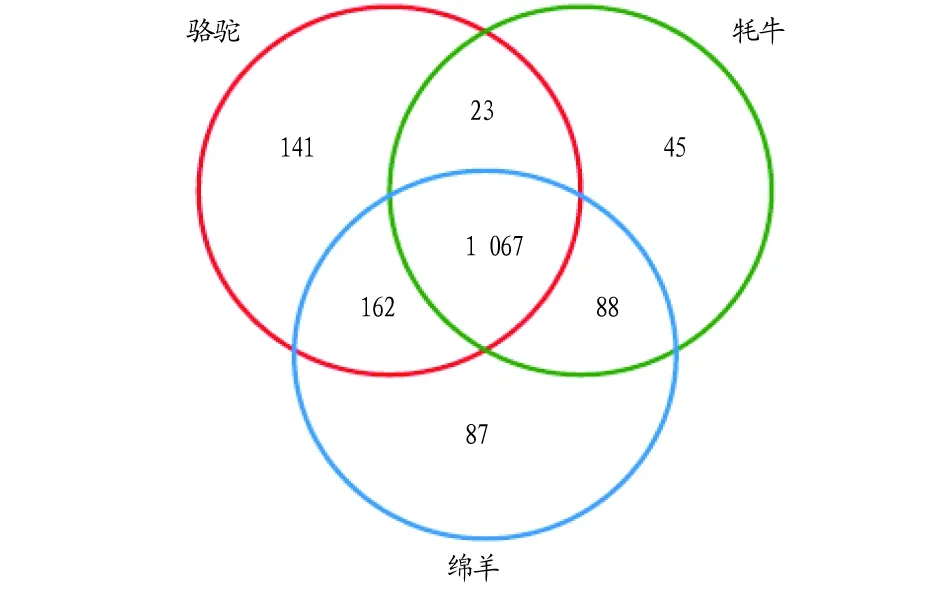
图2 不同动物肠道微生物种类的比较 Fig.2 Comparison of intestinal microbe populations in different animals
3种基因组中存在大量的高质量序列并未匹配到以上任何属中,这些序列没有与任何生物类群匹配,意味着这些序列与当前生物信息数据库中任何已知生物的序列没有相关性或者这些序列与它们的亲缘关系非常远。因此,这3种动物肠道中蕴藏着惊人的生物多样性。
为进一步了解不同动物之间的差异,根据COG之间的相似度关系,得到聚类后的菌群相对丰度比较热图。从图4可以看出,3种动物间COG具有较高的相似性。
2.4KEGG功能注释利用BLAST将基因集序列与KEGG 的基因数据库(GENES)进行比对,将比对结果使用KOBAS 2.0软件进行功能注释。使用KO、Pathway、EC、Module 对应的基因丰度总和计算该功能类别的丰度。结果发现,骆驼、绵羊及白牦牛这3种动物肠道菌群可能参与的代谢通路分别为311、199及305个,涉及到各个方面,包括脂肪酸的代谢、多糖的代谢、蛋白酶以及肽酶的产生等。同时,3种动物共同所涉及的代谢通路有188条,因此3种动物肠道菌群所涉及的功能具有较高的相似性。
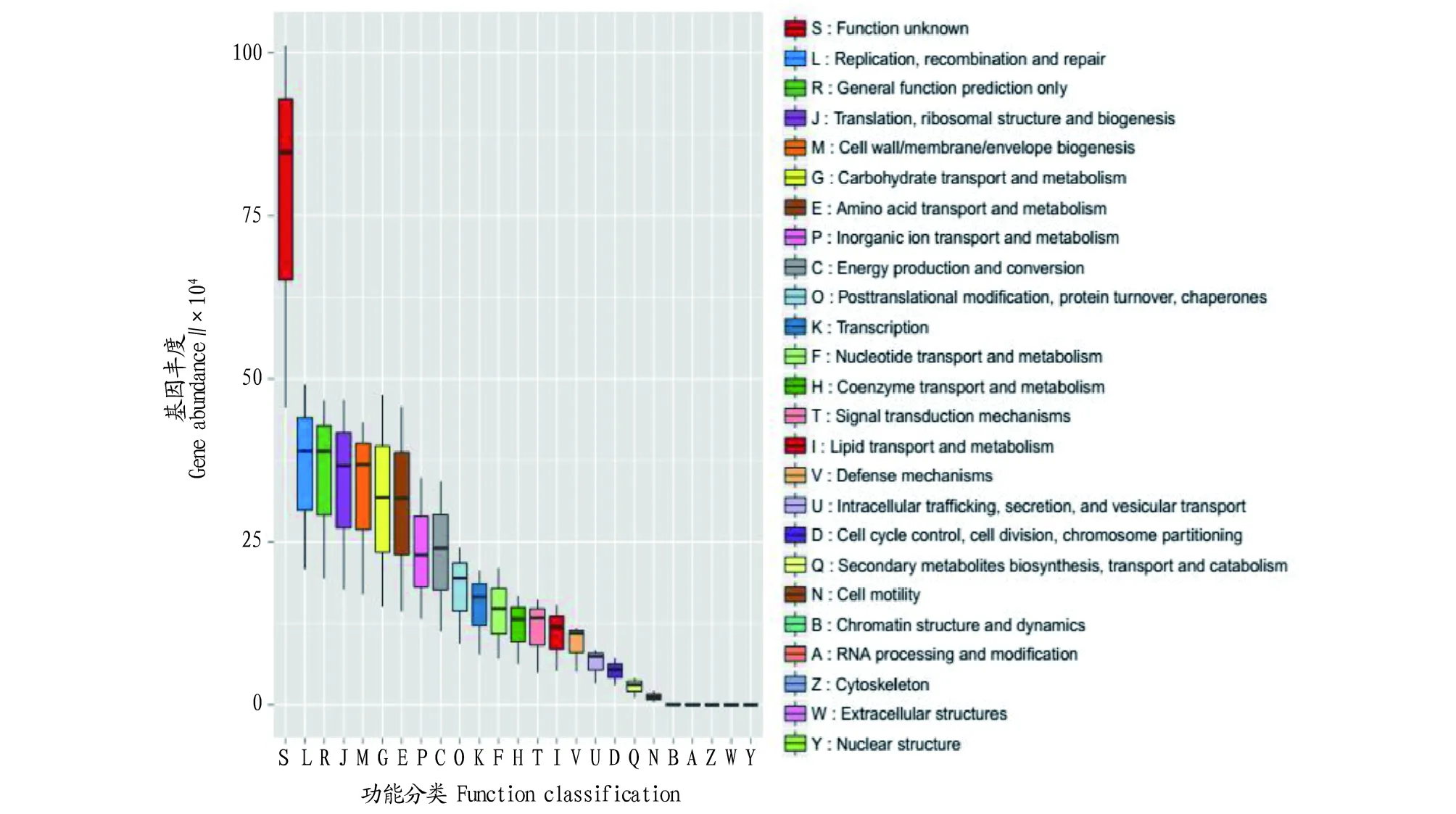
图3 不同动物肠道微生物种类的比较Fig.3 Comparison of intestinal microbe species in different animals
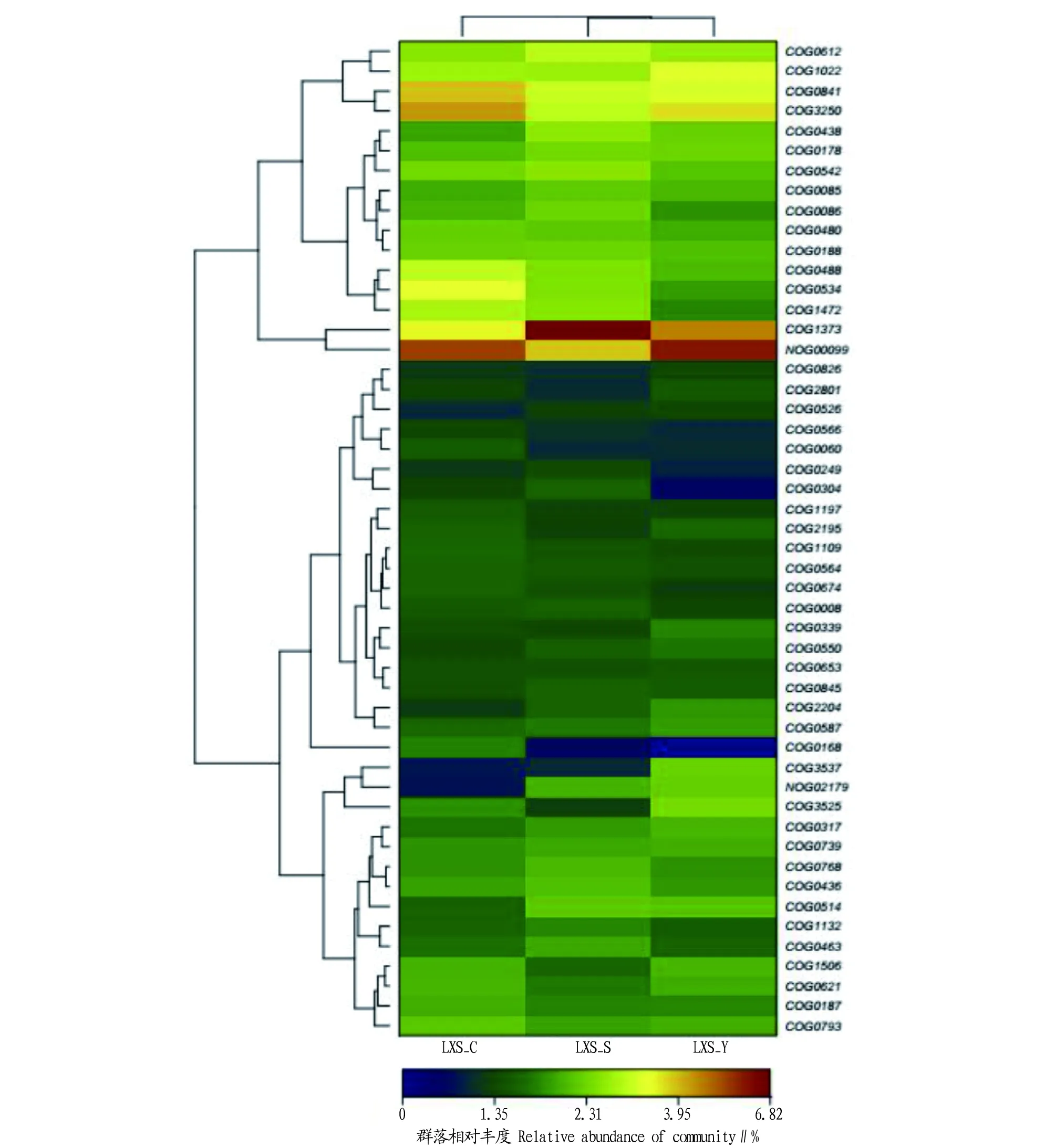
图4 不同动物肠道段及粪便肠道微生物属比较热图Fig.4 Heatmap analysis of COGs of genes in the intestinal segments and fecal intestinal microflora of different animals
3 讨论
已有的动物肠道微生物研究显示不同动物之间在肠道微生物多样性方面有明显差异。此外,环境在很大程度上影响动物肠道微生物多样性[8]。该研究结果也表明,骆驼、绵羊及白牦牛这3种动物肠道菌群是不同的,但由于其生存环境的一致性导致其肠道段微生物的种类和分布具有高度的相似性,这与以前的研究报道相一致。
不同动物的肠道段微生物的种类和分布特点和规律不同,在门水平上,拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、纤维杆菌门(Fibrobacteres)、变形菌门(Proteobacteria)、螺旋体门(Spirochaetes)、疣微菌门(Verrucomicrobia)和广古菌门(Euryarchaeota)等菌门为优势菌门,而在在属水平上拟杆菌属(Bacaeroides)、Alistipes、普氏菌属(Prevotella)和 纤维杆菌属(Fibrobacter)等菌属为优势菌属,具有明显的差异。此外,差异微生物所富集的代谢通路在不同动物中是不同的,主要差异在物质代谢和传染性疾病相关通路,但仍具有高度的相似性。
我国西部高海拔地区物种分布广泛、种类众多,该研究结果可为了解我国西部高海拔地区的微生物多样性,特别是我国所特有的白牦牛等动物的肠道微生物多样性提供重要信息。
