吡拉西坦片的处方工艺研究及溶出一致性评价
2019-04-03于曜荧陈玉双郑玉林孙长山
于曜荧,杨 静,陈玉双,郑玉林,孙长山*
吡拉西坦片的处方工艺研究及溶出一致性评价
于曜荧1,杨 静2,陈玉双2,郑玉林2,孙长山1*
(1. 沈阳药科大学 药学院,辽宁 沈阳 110016;2. 上海峰林生物科技有限公司,上海 201203)
以溶出曲线相似性2为指标,筛选吡拉西坦片处方工艺,并进行放大验证。开发适宜的吡拉西坦片溶出曲线测定方法,采用沸腾制粒工艺制备吡拉西坦片,采用HPLC法对片剂进行溶出含量测定,对比自制制剂与原研制剂在不同溶出介质中的溶出行为。自制制剂与原研制剂体外溶出一致,处方工艺可行。吡拉西坦片体外溶出曲线一致性评价合格,并且该制备工艺简单,质量可控,工艺适应性强。
药剂学;一致性评价;处方工艺;溶出曲线;HPLC;吡拉西坦
吡拉西坦片(脑复康片),临床上用于急、慢性脑血管病、脑外伤、各种中毒性脑病等多种原因所致的记忆减退及轻、中度脑功能障碍的治疗,也可用于儿童智能发育迟缓等症。本品为脑代谢改善药,属于-氨基丁酸的环形衍生物。有抗物理因素、化学因素所致的脑功能损伤的作用。能促进脑内ADP转化为ATP,可促进乙酰胆碱合成并正增强神经兴奋的传导,具有促进脑内代谢作用。可以对抗由物理因素、化学因素所致的脑功能损伤。
药物一致性评价,最早是《国家药品安全“十二五”规划》中提出的一项药品质量要求,即国家要求仿制药品要与原研药品质量和疗效一致。具体来讲,要求杂质谱一致、稳定性一致、体内外溶出规律一致。开展仿制药质量和疗效一致性评价工作,对提升我国制药行业整体水平,保障药品安全性和有效性,促进医药产业升级和结构调整,增强国际竞争能力,都具有十分重要的意义。
本品为医保乙类药品,临床应用较为广泛,是经临床验证有效并有很好耐受性的药物。按照国家目前相应的政策法规要求,吡拉西坦片需要进行质量与疗效一致性评价研究。本文作者采用流化床制粒工艺制备片剂,开发出适宜的溶出方法,并与原研制剂进行体外溶出行为的比较,进行一致性评价,验证处方及工艺的可行性。
1 仪器与试药
GHL-2.5湿法制粒机、FLP-0.5流化床制粒机(常州佳发制粒干燥设备有限公司),VH-5混合机(上海天峰制药设备有限公司),ZP-10A旋转式压片机(北京新龙立科技有限公司),LDCS高效包衣机(日本Freund-Vector公司),Ultima IV型X射线粉末衍射仪(日本理学株式会社),DSC3差示扫描量热仪(梅特勒托利多精密仪器有限公司),Agilent 1260高效液相色谱仪(美国Agilent公司),RC806D溶出试验仪(天津天大天发科技有限公司),pH1801酸度计(梅特勒-托利多仪器(上海)有限公司),BSA224S-CW电子天平(德国Sartorius科学仪器有限公司),EX125DZH电子天平(奥豪斯仪器(常州)有限公司)。
吡拉西坦片(UCB Pharma GmbH,商品名为Nootrop,规格800 mg,批号 222686,208375),吡拉西坦对照品(含量质量分数为100%,中国食品药品检定研究院,批号100386-200703),交联羧甲基纤维素钠(批号170902)、无水胶态二氧化硅(批号171001)(安徽山河药用辅料股份有限公司,PEG6000(南京威尔药业股份有限公司,批号20160901s-2),硬脂酸镁(辽宁奥达制药有限公司,批号20170202),Y-1-7000型欧巴代(卡乐康,批号THL48744),OY-S-29019欧巴代(上海卡乐康包衣技术有限公司,批号THL51367),乙腈(德国Merk公司,色谱纯,批号UN1648),磷酸氢二钾(德国CNW科技公司,色谱纯,批号E0830012),其他试剂(分析纯,市售),水为纯化水(自制)。
2 方法与结果
2.1 吡拉西坦片的处方工艺
2.1.1 原辅料的相容性试验
根据资料检索[1],吡拉西坦片原研制剂处方中的辅料种类为:PEG6000、无水胶态二氧化硅、交联羧甲基纤维素钠、硬脂酸镁、羟丙甲纤维素、二氧化钛和PEG400。经处方分析,后三者为包衣成分,包衣粉具体型号为欧巴代Y-1-7000及欧巴代OY-S-29019。
参照指导原则[2],将吡拉西坦原料与上述各辅料按照比例要求混合,分别进行光照、高温、高湿试验,考察各样品原料药的含量、有关物质、晶型等项目各时间节点的变化。结果显示,相关考察项目与参比制剂相比,无明显差异。故原辅料相容性良好,可以按照上述辅料进行处方摸索。
2.1.2 原料药晶型的确定
吡拉西坦共报道了[3-8]五种晶型和两种水合物:晶型I~晶型Ⅴ、一水合物(piracetam monohydrate)、二水合物(piracetam dihydrate)。其中晶型Ⅳ和晶型Ⅴ只能在压力大于0.5 GPa才能制得;晶型Ⅰ是通过将晶型Ⅱ或者晶型Ⅲ升温至127 ℃然后迅速降至室温下制得,但晶型Ⅰ极不稳定,在室温下即迅速发生转晶;室温下,晶型Ⅱ是亚稳晶型,晶型Ⅲ是稳定晶型。在溶剂介导下,晶型Ⅱ会转变为晶型Ⅲ。
通过对参比制剂、原料药及自制样品的XRD和DSC检测分析,并结合文献图谱,经对比,表明原料药和自制样品和参比晶型相同,为晶型III。检测图谱见图1~4。
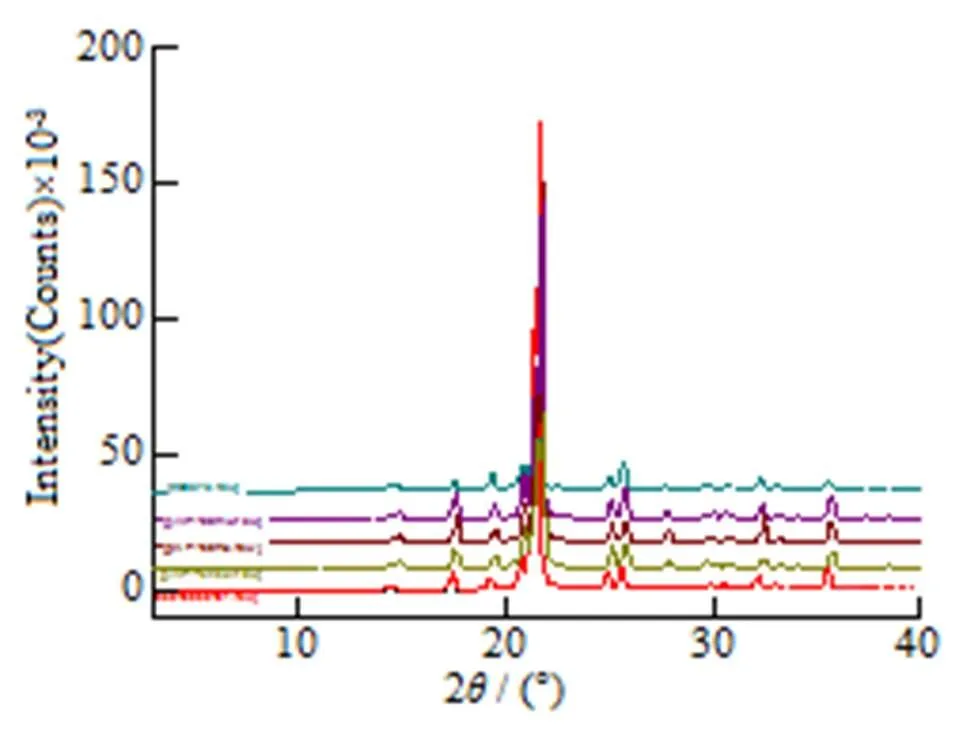
Fig. 1 XRD pattern of piracetam and reference
图1 原料药与参比制剂XRD图谱
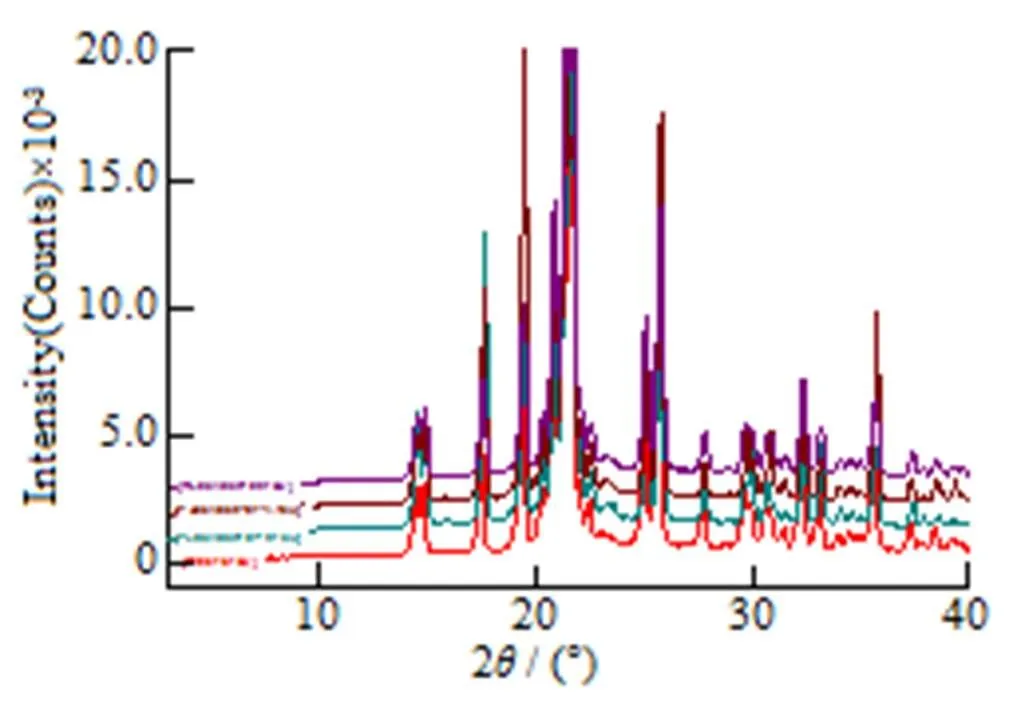
Fig. 2 XRD pattern of homemade and reference
图2 自制制剂与参比制剂XRD图谱
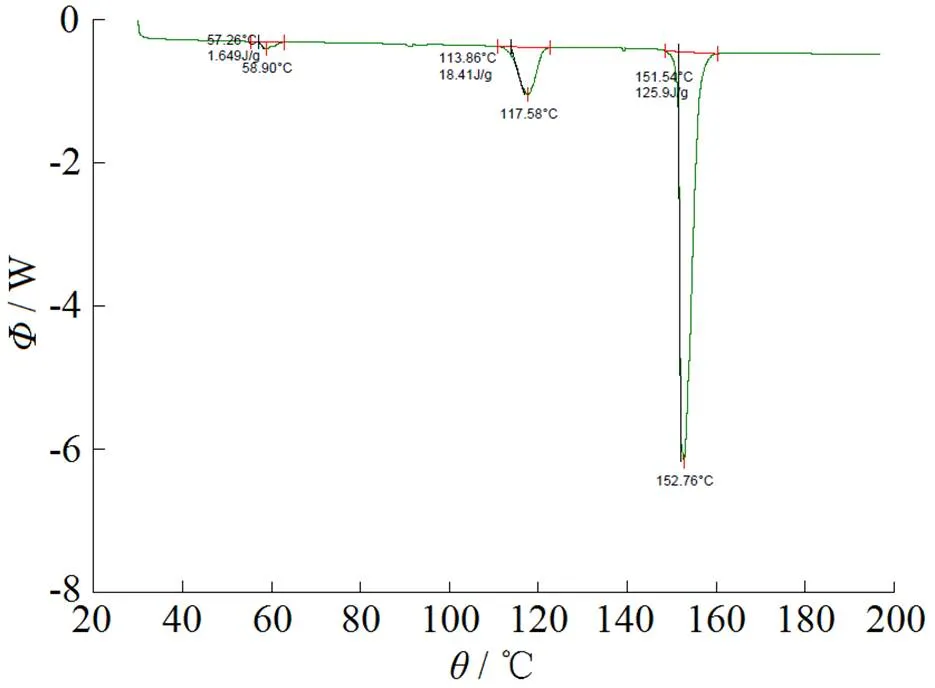
Fig. 3 DSCatlas of reference
图3 参比制剂DSC图谱
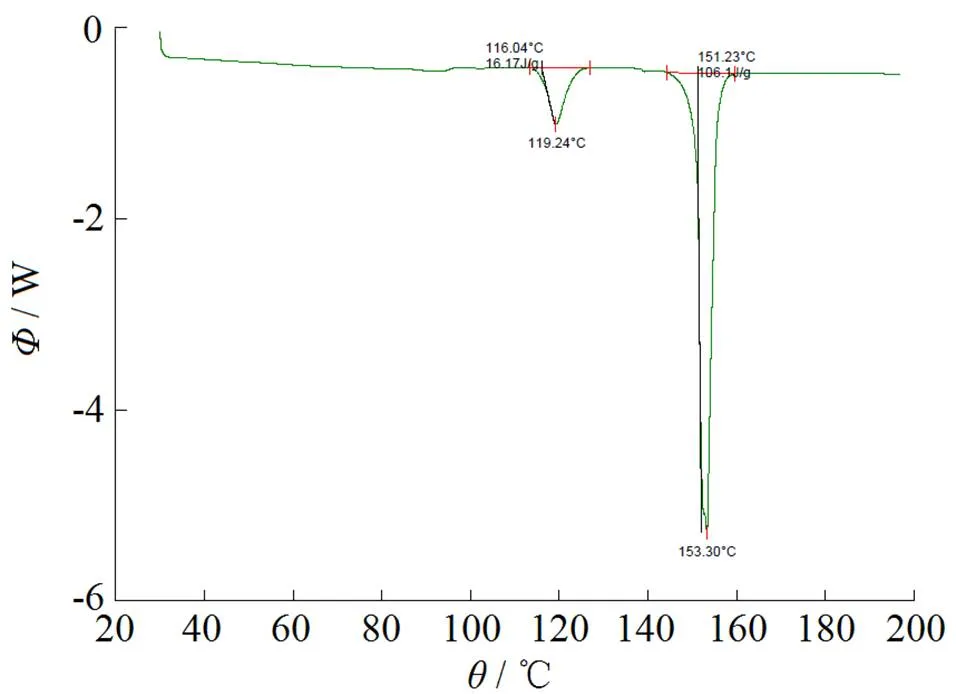
Fig. 4 DSC atlas of homemade
2.1.3 处及制备工艺的筛选
根据原研的辅料种类设计以下处方(表1),分别考察崩解剂的量及加入方式、制粒工艺和其他辅料量。先将吡拉西坦原料药粒径控制在200 μm以下备用,按照处方量(800片)称量原辅料,分别以纯化水为黏合剂,采用湿法制粒及沸腾制粒的工艺进行制粒,干燥、混合,用11 mm浅凹圆冲压片,硬度10~15 kg。考察指标及检测结果(表2)。由结果可知,处方1、2、3用水湿法制粒,颗粒黏性较差,片剂溶出较快;处方4加大硬脂酸镁用量,片剂溶出变慢,虽2较好,但总体溶出量会变小;处方5、6分别加大二氧化硅及PEG6000用量,片剂溶出会变快;综合考量处方7较好,并与原研制剂在pH值1.2介质中进行溶出2比较,结果2=59(2>50),较原研制剂溶出偏快,目的是为包衣留出下降空间。筛选后具体处方如下(每片):吡拉西坦400 mg、PEG6000 8.35 mg、无水二氧化硅7.35 mg、交联羧甲基纤维素钠10 mg、硬脂酸镁2 mg,理论片质量427.7 mg。
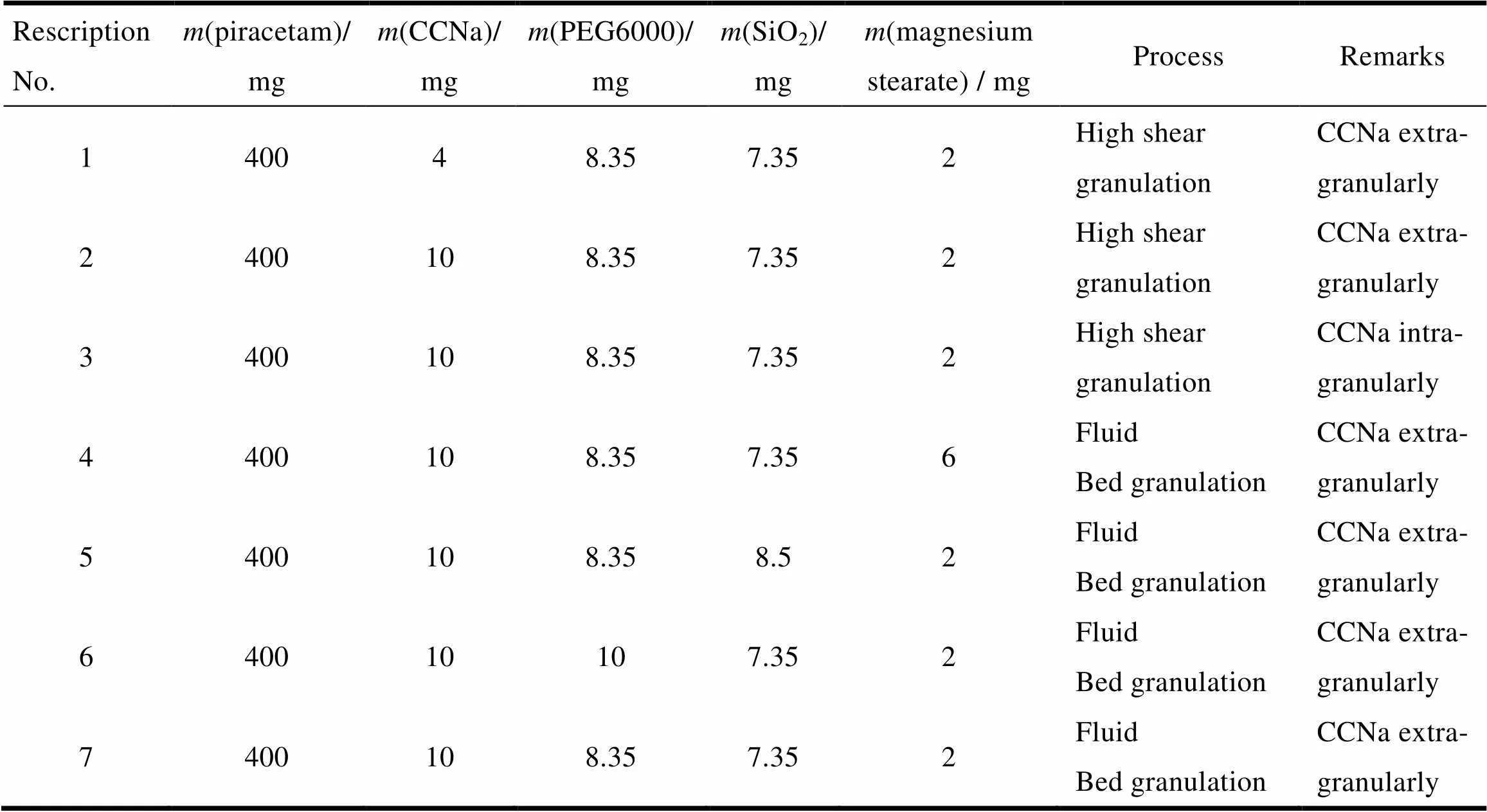
Table 1 The table of prescription screening
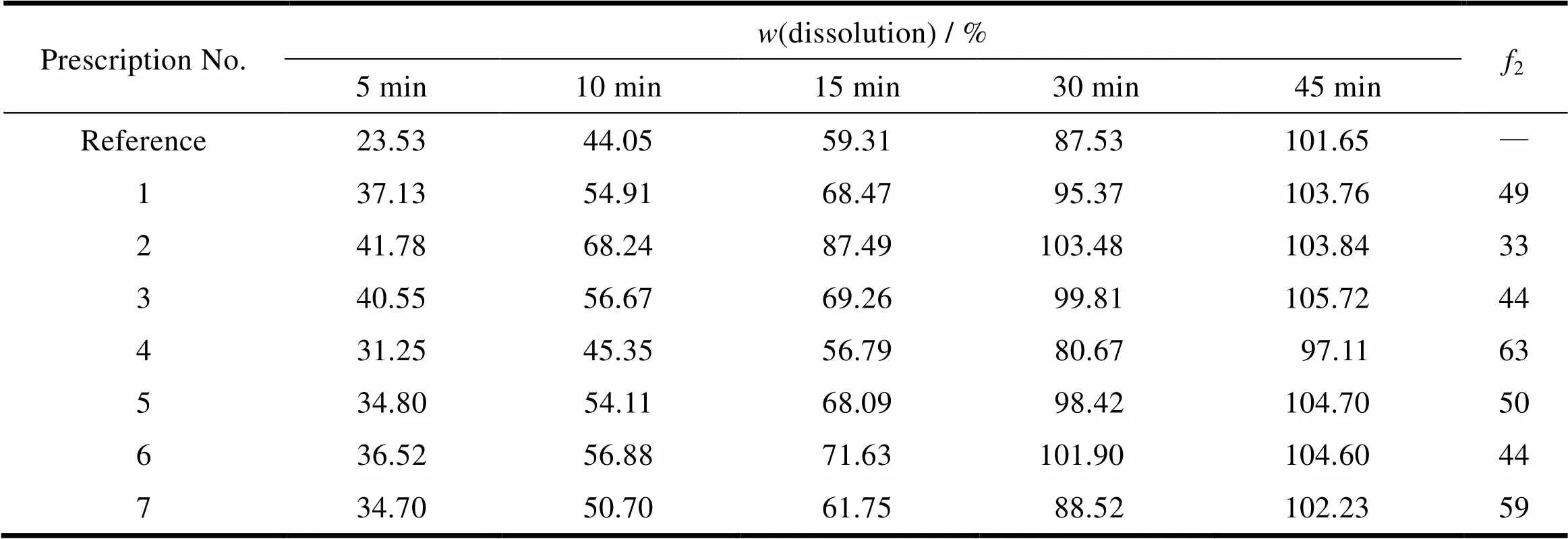
Table 2 The dissolution results in pH 1.2 medium
2.1.4 包衣工艺的考察
吡拉西坦片包衣粉型号为欧巴代Y-1-7000及欧巴代OY-S-29019,即需要包两层薄膜衣。经成分分析,欧巴代Y-1-7000含二氧化钛遮光成分,其作用是遮光及防潮,欧巴代OY-S-29019的作用是抛光。内层薄膜衣分别考察理论质量增重1%、2%和3%对外观及药物溶出的影响,结果见表3。外层抛光衣分别考察理论质量增重0.1%及0.7%,结果见表4。从结果可知,内层包衣对片剂的溶出基本上无影响,而外层包衣影响较大,随着包衣增重的增大溶出变慢。经综合考察,分别选择3%和0.7%的包衣增重。
包衣液的配制:取胃溶型薄膜包衣预混剂(欧巴代Y-1-7000)及水适量,按比例配制成质量分数为15%固含量的包衣液,搅拌45 min,备用。取胃溶型薄膜包衣预混剂(欧巴代OY-S-29019)及水适量,按比例配制成质量分数3%固含量的包衣液,搅拌使全部溶解成澄清透明液体,备用。
包衣工艺:根据设备情况调整喷枪与片床距离、进风温度、雾化压力和流速等参数;待片床温度达到40 ℃时,打开喷枪、蠕动泵,开始包内层薄膜衣(质量分数15%的欧巴代Y-1-7000水溶液),通过调节进风温度,保持片床温度在38~42 ℃内,待理论增重达到3%时,停止喷液,干燥2.0~3.0 min;继续包外层抛光衣(质量分数3%的欧巴代OY-S-29019水溶液),参数同上,包衣结束后干燥2.0~3.0 min。
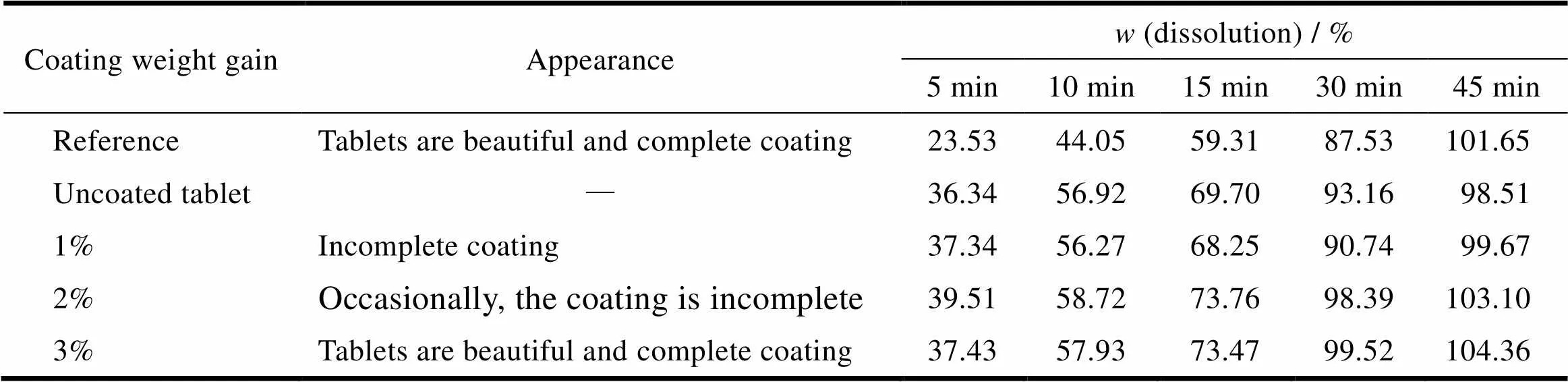
Table 3 The table of inner coating weight gain screening in pH1.2medium
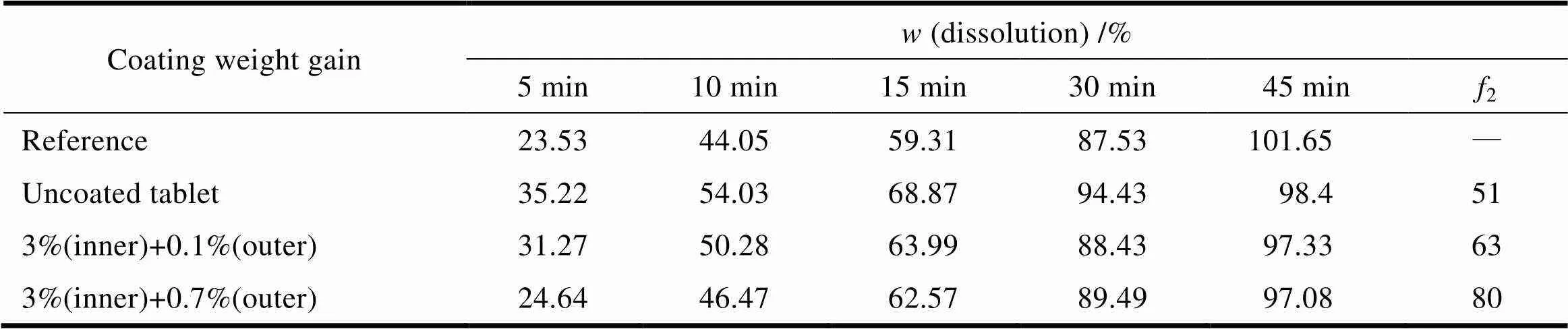
Table 4 The table of outer coating weight gain screening in pH1.2 medium
2.1.5 中试放大研究
为考察处方及工艺的适用性,对该工艺进行放大研究,批量为50万片。流化床制粒工艺:以纯化水为黏合剂;风机频率30~50 Hz,进风温度(80±10) ℃,物料温度(40±5) ℃,雾化压力0.3~0.5 MPa,蠕动泵转数100~150 r·min-1;将粒径在200 μm以下的吡拉西坦原料药真空抽进到制粒机中,按上述参数进行制粒,制粒后干燥至水分≤2%,以 500 μm筛整粒。制粒后按照处方量分别加入交联羧甲基纤维素钠、PEG6000和二氧化硅及硬脂酸镁,混合均匀,压片,包衣。样品检测结果见“2.3”条。
2.2 溶出方法的开发与验证
经检索各国药典,吡拉西坦片只有《中华人民共和国药典》收载,但未考察溶出度,故需新建立具备适宜区分力的溶出方法。
2.2.1 溶出含量测定方法的筛选
分别对比《中华人民共和国药典》2015版二部中方法[9]与《欧洲药典》中原料含量方法[10],后者经微调,四介质中吡拉西坦与相邻峰均分离良好,结果见图5。分离效果后者优于前者,因此选用后者方法作为最终的溶出分析方法,即:流动相为乙腈-1.0 g·L-1磷酸氢二钾溶液(体积比10∶90),稀磷酸调节至pH值6.0;流速为1.0 mL·min-1;色谱柱为十八烷基键和硅胶柱(25 cm×4.6 mm,5 μm);进样量10 μL;检测波长210 nm。
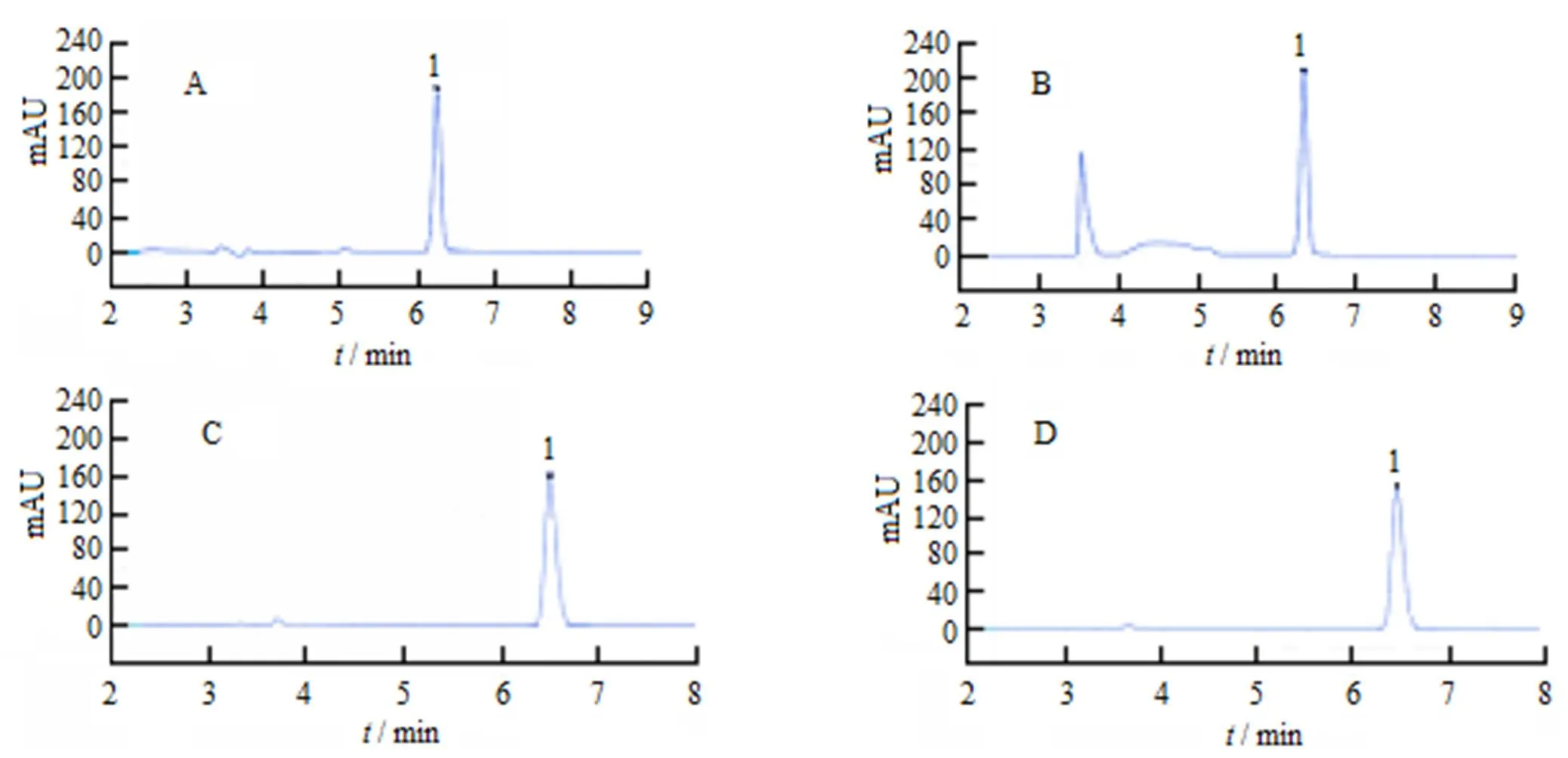
Fig. 5 Chromatograms of pH 1.2medium(A), pH 4.0 medium(B), pH 6.8 medium(C) and water medium(D)
2.2.2 吡拉西坦溶解度的测定
分别在不同pH值溶液中加入过量吡拉西坦原料药,在37 ℃恒温震荡,24 h后参照“2.2.1”条测定方法注入液相色谱中,记录色谱图,计算含量,结果见图6。由图6可见,吡拉西坦除在无离子强度的水中溶解度大之外,在其他介质中无明显pH依赖,故溶出介质选择常用的pH值1.2、pH值4.0、pH值6.8和水。
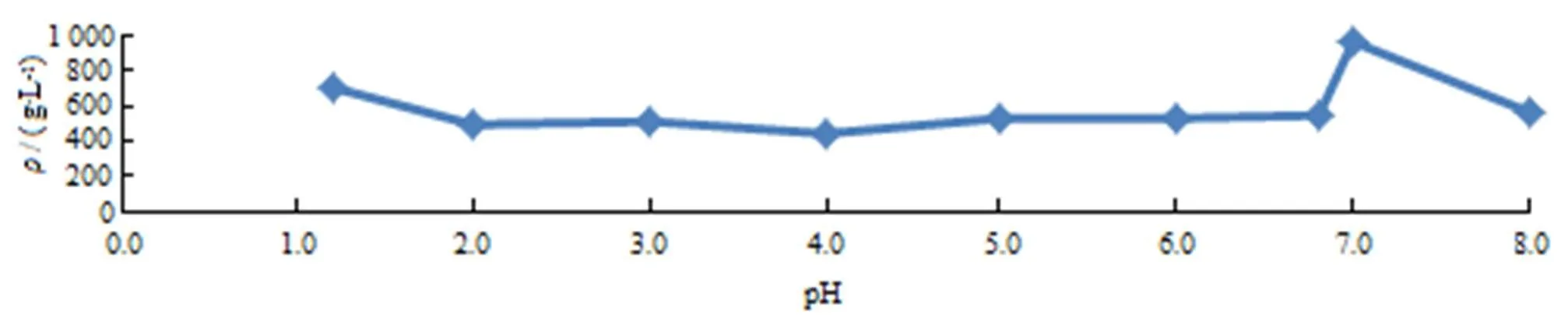
Fig. 6 Thesolubility ofpiracetam in different pH media
2.2.3 溶出方法的筛选与建立
固体制剂体外溶出评价中,开发适宜区分力的溶出曲线测定方法尤为重要,溶出方法包括桨法、篮法,溶出参数包括转数、介质体积。故通过测定参比制剂在各方法及参数条件下的溶出数据,比较选出适宜的溶出方法。
通过筛选,在篮法75 r∙min-1、900 mL条件下,参比制剂在各介质中各时间点的RSD均符合指导原则规定,并有一定的区分力。篮法75 r·min-1相当于桨法50 r·min-1,能够模拟老年人胃肠蠕动情况,指导原则推荐以低转速为起始研究点,因此作者选择篮法作为参比制剂和自制制剂比较的方法。
2.2.4 滤膜吸附性验证
将滤膜在纯化水中浸泡24 h,对本品片剂在各介质中的溶出液进行过滤,以高速离心处理的同介质上清液中的吡拉西坦含量作为100%,考察吸附性,结果见表5。在4种介质中,供试品在过滤前后的含量基本上没有差异,其质量分数均小于1%,即水相、聚醚砜膜对吡拉西坦无吸附。
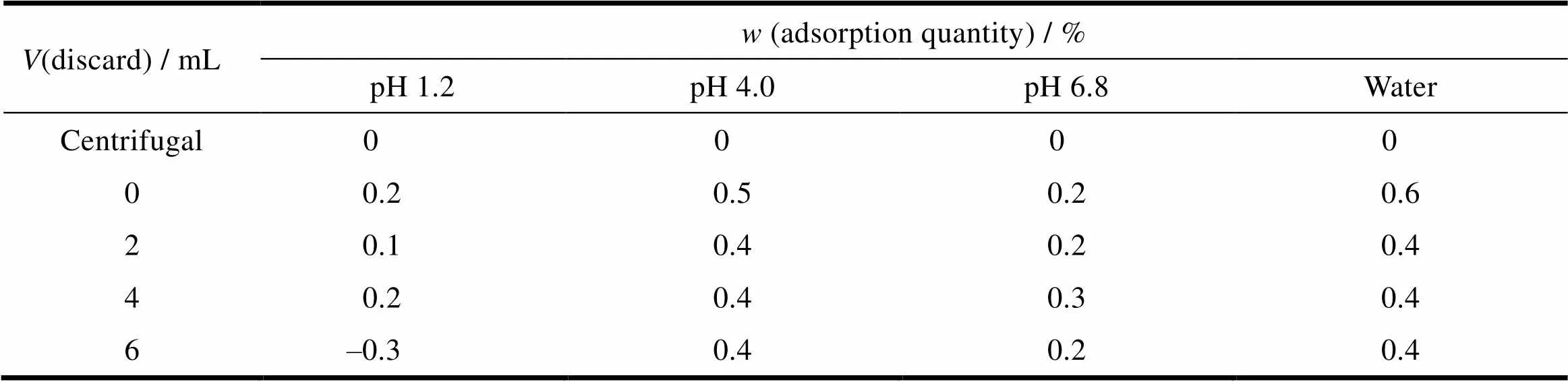
Table 5 Results of membrane adsorption test
2.2.5 溶液稳定性验证
取本品片剂,分别考察各介质的溶出液在室温及溶出条件(37 ℃)下,不同时间样品溶液含量,结果见表6。由表6可知:(1)吡拉西坦供试溶液室温条件下,水介质、pH值4.0介质、pH值6.8介质48 h溶液稳定,pH值1.2介质中室温下8 h溶液稳定,24 h存在降解,该介质样品测定需在8 h内完成。(2)吡拉西坦供试溶液37 ℃水浴条件下,4种介质2 h内均稳定,可保证溶出度测定准确。
2.2.6 准确度试验
称取一定量吡拉西坦原料药,分别稀释至片剂溶出量的50%、80%、100%(即44.4、71.1、88.9 mg∙L-1),加入处方量的空白辅料,每个质量浓度配制3个样品,每个样品进2针注入液相色谱仪,以外标法计算本品在4种介质中的回收率,结果见表7。
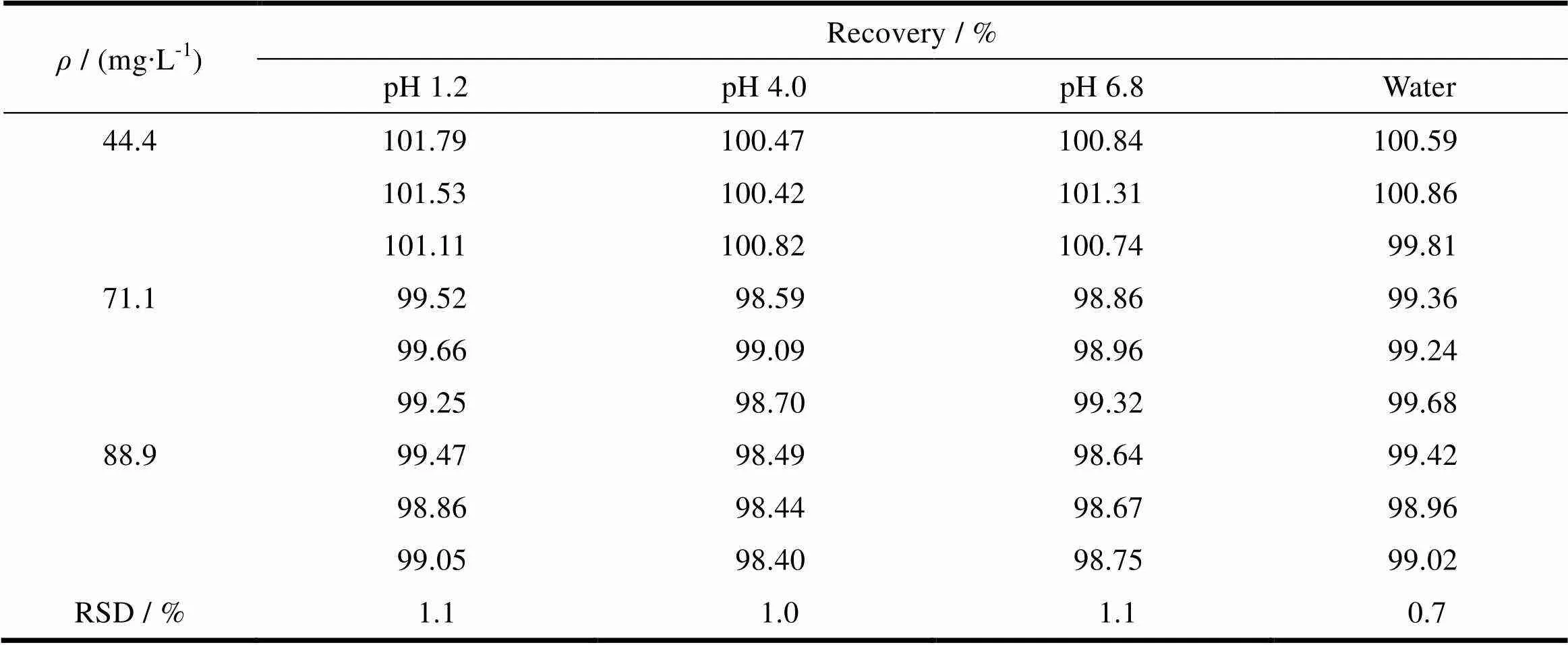
Table 7 Results of recovery of piracetam
结果表明,本品在不同介质中的回收率为98.0%~102.0%,RSD均<2%,可满足本品在不同介质中溶出度测定的要求。
2.2.7 重复性试验
取本品片剂5片,研磨后称取约20 mg,分别用四介质稀释至相当于溶出量的100%,检测片剂含量,平行配制6个样品,注入液相色谱仪中,计算含量,结果见表8。
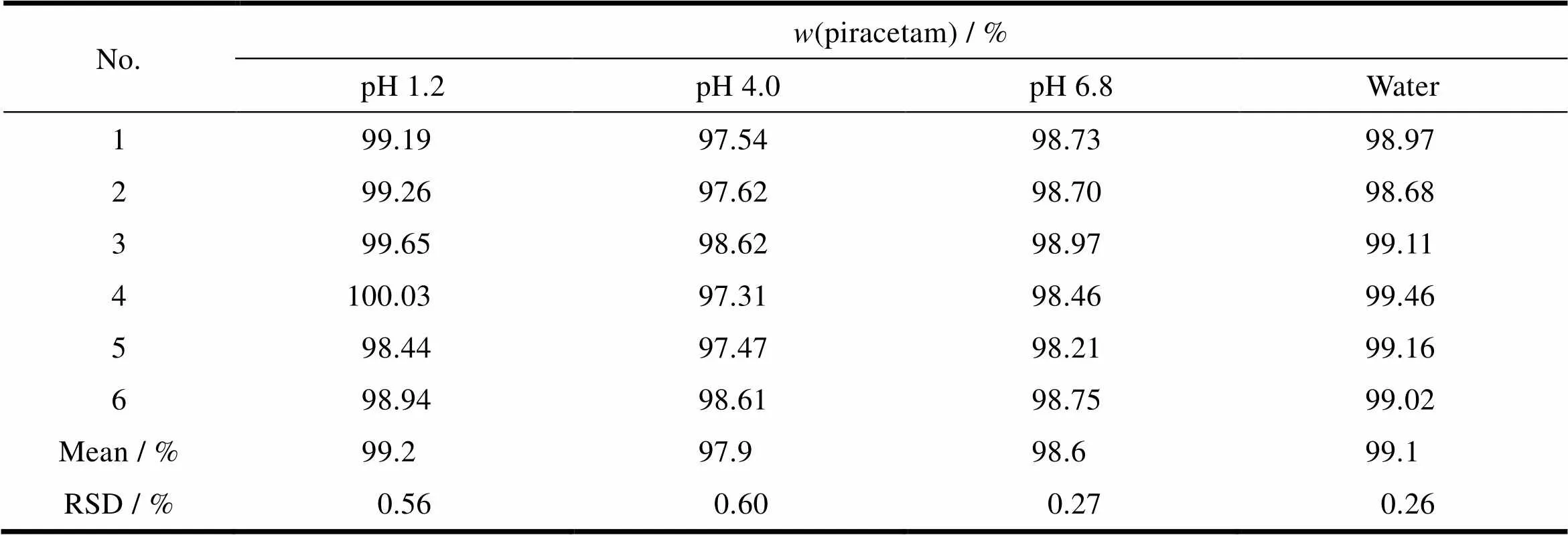
Table 8 Results of reproducibity of piracetam
结果表明,分别在不同介质中测定本品含量的RSD<1%,重复性较好,可满足本品溶出度测定的要求。
2.2.8 线性关系试验
取吡拉西坦对照品,配制成一定质量浓度的母液,用移液管吸取不同体积稀释至质量浓度为11~113 mg∙L-1的溶液,即相当于溶出量的12.5%~125%,分别注入液相色谱仪,记录色谱图。以质量浓度(mg∙L-1)为横坐标,峰面积()为纵坐标,绘制标准曲线,结果见图7。结果显示,吡拉西坦在四种介质中,溶液质量浓度在11~113 mg∙L-1内呈良好的线性关系。线性方程如下:(1)pH值1.2介质=14.032+11.593,2=0.999 3;(2)pH值4.0介质=14.11+9.213,2=0.999 9;(3)pH值6.8介质=14.156+7.201,2=1.000 0;(4)水介质=14.32+2.103,2=0.999 9。

A—pH 1.2 medium; B—pH 4.0 medium; C—pH 6.8 medium; D—Water medium
Fig. 7 The standard curve of piracetam
图7 吡拉西坦质量浓度与峰面积线性关系图
2.3 体外溶出曲线的评价
因国内吡拉西坦片上市规格均为0.4 g,而参比制剂规格为0.8 g,根据一致性评价改规格研究相关指导原则[11],需以相同剂量给药,故溶出曲线重点研究及测定2片自制样品。
取自制中试样品2片与参比制剂1片分别在pH值1.2、pH值4.0、pH值6.8及水4种介质中进行溶出度比较,采用2相似因子法对自制片和参比片进行溶出度评价,在4种介质中2均大于70,故判定溶出一致。溶出曲线见图8。
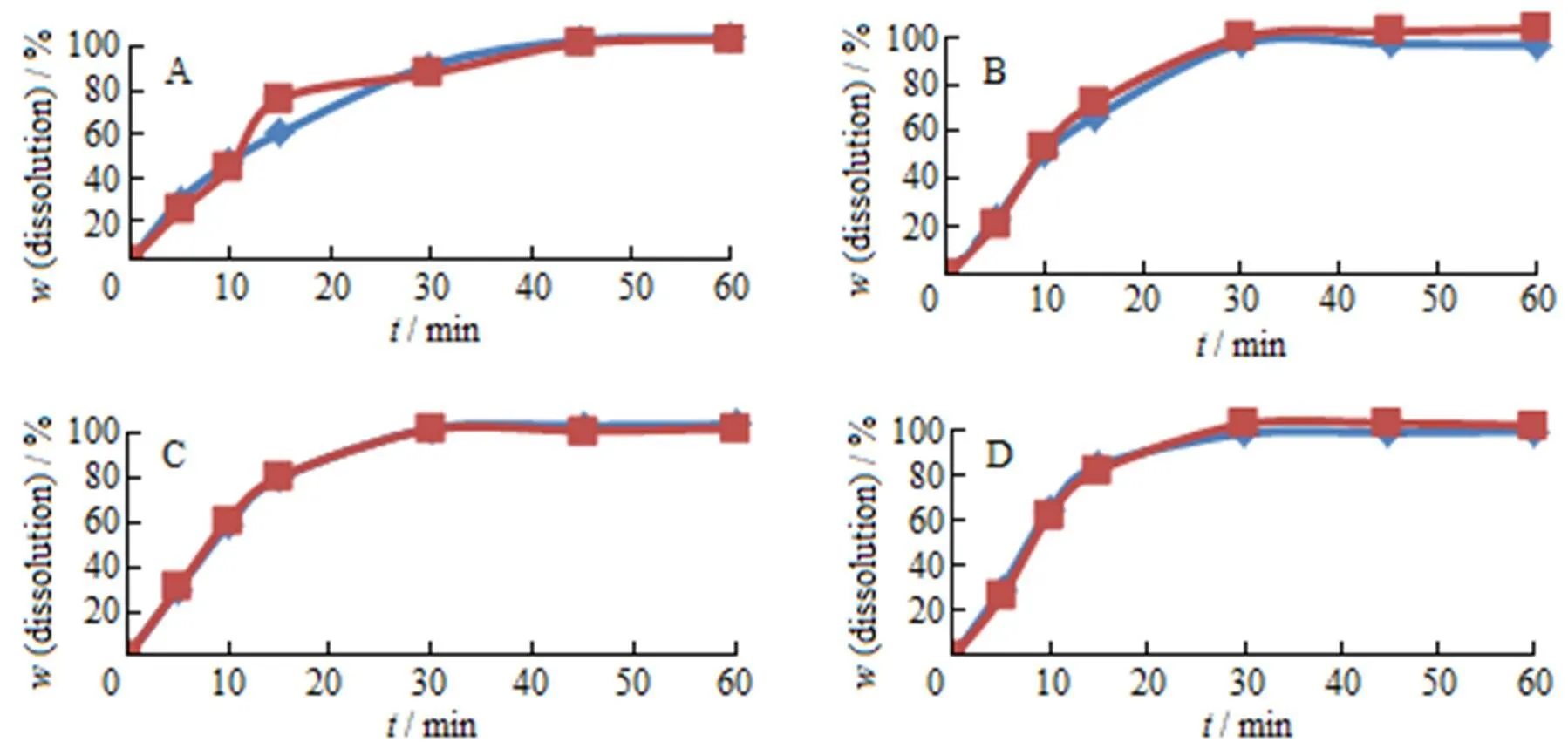
A—pH 1.2 medium; B—pH 4.0 medium; C—pH 6.8 medium; D—Water medium;·—Homemade; ¾—Reference
3 讨论
a. 吡拉西坦片溶出度的测定采用HPLC法,如采用紫外分光光度法,溶出四介质(pH值1.2、pH值4.0、pH值6.8、水)的空白样品在210 nm的检测波长下均有较强的末端吸收,会影响吡拉西坦含量的检测,因此采用液相方法测定吡拉西坦片溶出度。
b. 对于固体制剂,若想使体外溶出试验尽可能反映其在体内的释放和吸收情况,选择合适的、具有较强区分能力的溶出介质非常重要。本研究通过对桨法、篮法及介质体积、转数等参数在4种介质中溶出结果的对比研究,选定了该溶出方法。该方法在符合各时间点RSD规定的同时,有着较好的区分力,可以更好的评价自制制剂与参比制剂的体外差异。
c. 吡拉西坦片的质量与疗效一致性评价工作属于改规格药品研究。根据指导原则,对于改规格药品“建议以改规格药品和参比制剂以相同剂量给药(单次给药剂量不超过最大给药剂量)进行生物等效性试验”。故药学研究中,体外溶出曲线重点研究及测定2片自制样品与参比制剂1片进行比较。
4 结论
吡拉西坦片目前质量标准均未考察溶出度及溶出曲线,药品质量及标准均有待提高。作者通过对处方工艺摸索及溶出方法开发,制得的吡拉西坦片与原研片剂辅料组成一致,体外溶出行为一致,并且制备方法简单,质量可控,适合工业化生产。
[1] UCB S A. 吡拉西坦片说明书. 阿根廷: 2011.
[2] 《化学药物制剂研究基本技术指导原则》课题研究组. 化学药物制剂研究基本技术指导原则[S]. 北京: 国家食品药品监督管理总局, 2005.
[3] O’MAHONY M A, MAHER A, CROKER D M, et al. Examining solution and solid state composition for the solution-mediated Polymorphic transformation of carbamazepine and piracetam[J]. Crystal Growth & Design, 2012, 12(4): 1925-1932.
[4] MAHER A, CROKER D, ÅKE C. Rasmuson, et al. Solubility of form III piracetam in a range of solvents[J]. J Chem Eng Data, 2010, 55(11): 5314-5318.
[5] MAHER A, SEATON C C, HUDSON S, et al. Investigation of the solid-State Polymorphic transformations of piracetam[J]. Crystal Growth & Design, 2012, 12(12): 6223-6233.
[6] PICCIOCHI R, DIOGO H P, DA P M. Thermodynamic characterization of three polymorphic forms of piracetam[J]. Journal of Pharmaceutical Sciences, 2011, 100(2): 594-603.
[7] FABBIANI F P A , ALLAN D R , DAVID W I F , et al. High-pressure studies of pharmaceuticals: An exploration of the behavior of piracetam[J]. Crystal Growth & Design, 2007, 7(6):1115-1124.
[8] CHAMBRIER M, BOUHMAIDA N, BONHOMME F, et al. Electron and electrostatic properties of three crystal forms of piracetam[J]. Crystal Growth & Design, 2011, 11(6): 2528-2539.
[9] 国家药典委员会. 中华人民共和国药典: 二部[M]. 北京: 中国医药科技出版社, 2015: 466-467.
[10] 欧洲药典委员会. Ph.Eur 9.0[M], 法国: 欧洲药品质量管理局, 2017: 3353-3354.
[11] 国家食品药品监督管理总局. 仿制药质量和疗效一致性评价工作中改规格药品(口服固体制剂)评价一般考虑[S]. 北京: 国家食品药品监督管理总局, 2017.
Study on the formulation process and dissolution consistency evaluation of piracetam tablets
YU Yaoying1, YANG Jing2, CHEN Yushuang2, ZHENG Yulin2, SUN Changshan1*
(1.,,110016,; 2.., L, S201023,)
To take the similarity2of dissolution curve as an index, the formulation process of piracetam tablets was screened and verified by amplification.A suitable method for determination of dissolution curve of piracetam tablets was developed. Piracetam tablets were prepared by boiling granulation process. The dissolution content of tablets was determined by HPLC. The dissolution behaviors of self-made preparation and original preparation in different dissolution media were compared.The self-made preparation was consistent with the original preparation indissolution, and the prescription process was feasible.Thedissolution profile of piracetam tablets is evaluated as conformance. And the preparation process is simple, the quality is controllable, and the process adaptability is strong.
pharmaceutics; consistency evaluation; formulation process; dissolution curve; HPLC; piracetam
2018-12-14
于曜荧(1985-), 男(汉族), 辽宁沈阳人,工程师, 主要从事药物制剂产业化研究,Tel. 024-43520517,E-mailydyaoying_2006@126.com;
孙长山(1968-),男(汉族), 辽宁沈阳人, 教授, 博士, 博士生导师, 主要从事药物制剂新技术与新剂型的研究与开发, Tel. 024-43520517, E-mail freescs@163.com。
(2019)02–0017–11
10.14146/j.cnki.cjp.2019.02.001
R94
A
(本篇责任编辑:赵桂芝)
