乳糖酸阿奇霉素注射液质量评价
2019-03-29侯金凤寇晋萍王光裕刘照振李珉车宝泉李文东
侯金凤 寇晋萍 王光裕 刘照振 李珉 车宝泉 李文东
(北京市药品检验所,中药成分分析与生物评价北京市重点实验室,北京 102206)
阿奇霉素为第二代大环内酯类抗生素,最早由克罗地亚Pliva公司研制合成,1988年由Sour Pliva公司率先在前南斯拉夫上市,后将产品转让给美国辉瑞,1990年在英国上市[1-2]。1996年原研口服剂型(商品名“希舒美”)在中国上市。由于阿奇霉素具有对酸较稳定、在机体组织中浓度高、半衰期长、对革兰阳性菌和厌氧菌均有强大的抗菌活性等优点[3],被临床广泛应用。目前原研厂家上市的主要剂型包括口服剂型和注射用阿奇霉素(柠檬酸盐),但无注射液剂型。国内药厂在仿制过程中将阿奇霉素与多种酸结合成盐,提高溶解性制成注射剂[4]。然而,由于研究基础薄弱,并未对改盐基改剂型后产品进行深入研究即上市。乳糖酸阿奇霉素即为其中一类,剂型包括注射用无菌粉末和注射用浓溶液。
对阿奇霉素药物的不良反应监测结果表明,阿奇霉素注射剂不良反应显著高于口服制剂,注射液的不良反应/事件显著高于粉针剂,可累及胃肠系统损害、皮肤及其附件损害、免疫功能紊乱、肝功能异常、中枢神经系统损害等,提示国产阿奇霉素注射剂的安全性问题应予以重点关注[5-7]。
2009年国家评价性抽验结果揭示,成盐剂类型、工艺水平、原(辅)料质量与注射用阿奇霉素产品的杂质谱密切相关,是影响产品质量的关键因素;盐酸等强酸盐产品质量及稳定性较差;柠檬酸等弱酸盐产品质量及稳定性相对较好;鉴于国产注射用阿奇霉素质量存在明显不足,建议尽快统一注射用阿奇霉素的质量标准,淘汰不合理的成盐工艺[8-9]。虽然从中国药典2010年版起,注射用阿奇霉素的质量标准已经有了大幅度的提升[10],一些不合理的成盐工艺产品也基本淘汰出市场,但乳糖酸阿奇霉素及注射剂仍在国内市场有销售,且基于多种原因,各生产企业仍执行各自的国家药品标准,未进行标准提高。乳糖酸阿奇霉素及注射剂的现行质量标准中,有关物质和含量测定项分别采用专属性差、灵敏度低的薄层色谱法和微生物检定法,因此存在较高风险。本文按照国家药品评价性抽验的研究思路[11],通过探索性研究揭示乳糖酸阿奇霉素及注射剂的问题及风险点,并对当前乳糖酸阿奇霉素注射液的质量状况进行评价,为乳糖酸阿奇霉素原料及注射剂处方与工艺的合理性评价提供依据。
1 仪器与试药
1.1 仪器
岛津20A高效液相色谱仪;Waters e2695高效液相色谱仪;Thermo Scientific Q Exactive高效液相色谱-高分辨质谱联用仪。
1.2 试药
阿奇霉素对照品(批号:130593-201303,纯度:94.4%),阿奇霉素系统适用性对照品(批号:130609-201504),由中国食品药品检定研究院提供。阿奇霉素的已知杂质J、Q、R、A、I、F、S和L对照品,均为北京理工大学提供。来自国内1家企业的注射用乳糖酸阿奇霉素和来自国内3家企业的7批次乳糖酸阿奇霉素原料。来自原研企业的5批次注射用阿奇霉素。来自国内1家企业的6批次乳糖酸阿奇霉素注射液均为2017年国家评价性抽验样品。磷酸氢二钾、磷酸二氢铵、氢氧化钠、氨水、盐酸、磷酸为分析纯,甲醇、乙腈为色谱纯,Merck公司产品。水为超纯水。
2 实验方法
2.1 标准检验
按照企业提供的国家药品标准(试行)YBH10722004乳糖酸阿奇霉素注射液标准进行法定检验。
2.2 探索性研究
根据标准检验结果及检验中发现的问题,以药品的安全、有效和质量可控为目标,结合文献调研对该品种的了解,对产品的有关物质、含量测定、pH等项目进行分析。
2.2.1 杂质谱研究
参考中国药典2015年版(ChP 2015版)中阿奇霉素的有关物质测定方法,建立了HPLC法测定乳糖酸阿奇霉素注射液中有关物质。并应用所建立的方法进行杂质谱相关研究。
(1)色谱条件:色谱柱:资生堂MGⅡ C18柱(4.6mm×250mm, 5μm);流动相:流动相A为磷酸盐缓冲液(取0.05mol/L磷酸氢二钾溶液,用20%的磷酸溶液调节pH值至8.2)-乙腈(45:55),流动相B为甲醇,线性梯度洗脱,梯度洗脱程序详见表1;柱温:30℃;流速:1.0mL/min;检测波长:210nm。
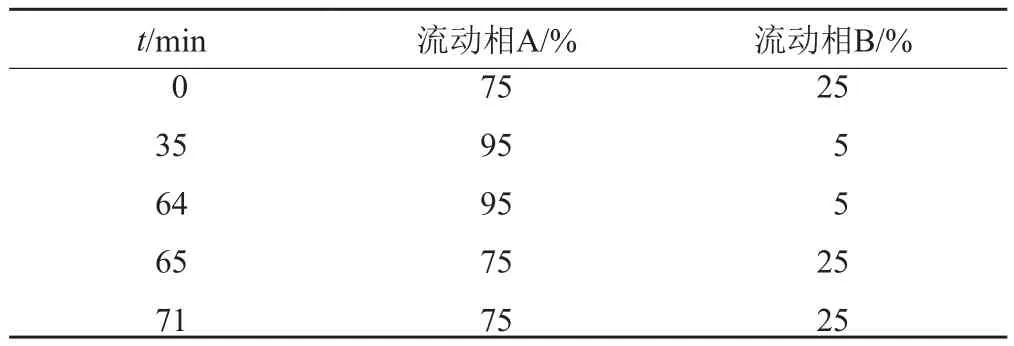
表1 HPLC梯度洗脱程序表Tab.1 HPLC linear gradient elution program
(2)强制降解试验:分别取乳糖酸阿奇霉素原料约1.0g,辅料约0.5g,阿奇霉素原料约0.5g及注射液10mL,按表2中条件制备样品溶液,分别进行热破坏后,采用“(1)”项中方法测定。

表2 强制降解试验样品制备Tab.2 Sample preparation for forced degradation test
(3)注射液贮藏条件对杂质影响的考察:取乳糖酸阿奇霉素注射液样品,冷藏于4℃冰箱中,放置3个月后,采用“(1)”项中方法测定有关物质含量。
(4)pH与有关物质相关性研究:模拟注射液的生产工艺,将溶液的初始pH值分别调整为5.5~7.5,制备样品溶液,在40和60℃条件下放置,进行加速试验,于第5天和第10天取出,采用“(1)”项中分析方法测定。
(5)未知杂质鉴定:质谱参数:电喷雾离子源(ESI),正离子模式检测,离子源温度:350℃;鞘气流速:35arb;辅助气流速:10arb;喷雾电压:3.5kV;毛细管:320℃;分辨率:70000;扫描模式:Full Scan(全扫描模式);扫描范围:100~1000m/z;S-Lens(离子传输透镜电压):50;加热温度:100℃;选择丰度较高的5强离子进行二级质谱分析,NCE(碰撞能量):25。称取阿奇霉素对照品约10mg,加甲醇使溶解并用甲醇稀释制成1μg/mL浓度的溶液,作为对照品溶液。取经HPLC分离后的未知杂质进行馏分收集、富集和纯化后,用甲醇溶解,作为供试品溶液。取以上对照品溶液和供试品溶液,采用流动注射分析法,获得一级高分辨数据及多级质谱数据。
(6)已知杂质活性研究:参考ChP2015版四部中阿奇霉素微生物检定法[12],采用抗生素检定用I号培养基(pH7.8~8.0),短小芽孢杆菌菌液为试验菌,pH7.8磷酸盐缓冲液稀释样品,以阿奇霉素标准品(10U/mL)为参照,对ChP 2015版中阿奇霉素项下[10]已知杂质J、Q、R、A、I、F、S和L的效价进行研究。考察各杂质在10、20、40、100、200和400U/mL 6个浓度水平下的抑菌活性。
2.2.2 含量测定
参考ChP2015版中注射用阿奇霉素的含量测定方法[10],建立了HPLC法测定乳糖酸阿奇霉素注射液的含量,色谱条件如下。色谱柱:资生堂MGⅡ C18柱(4.6mm×250mm, 5μm),流动相:磷酸盐缓冲液(取0.05mol/L磷酸氢二钾溶液,用20%的磷酸溶液调节pH值至8.2)-乙腈(45:55, V/V),等度洗脱,柱温:30℃;流速:1.0mL/min;检测波长:210nm。
3 结果与讨论
3.1 标准检验结果与分析
按现行质量标准检验,6批次样品均符合规定,合格率为100%。但出厂时间长的批次与新生产的批次相比,有关物质明显增加,含量明显降低,表明该制剂在有效期内不稳定,主成分不断降解,有关物质增加。此外,产品的pH值与出厂时间也呈明显相关性,随出厂时间的延长而降低,除1批次出厂时间较短的为6.8外,其余均小于6.0,而出厂时的pH值为7.4,pH频次分布图详见图1。
3.2 探索性研究结果
采用新建立的HPLC法测定产品中有关物质和含量,进而评价乳糖酸阿奇霉素注射液的质量及剂型的合理性。新建的HPLC方法可实现对产品中的全部杂质进行有效分离,检测限为0.1μg,定量限为0.3μg。
3.2.1 杂质谱分析
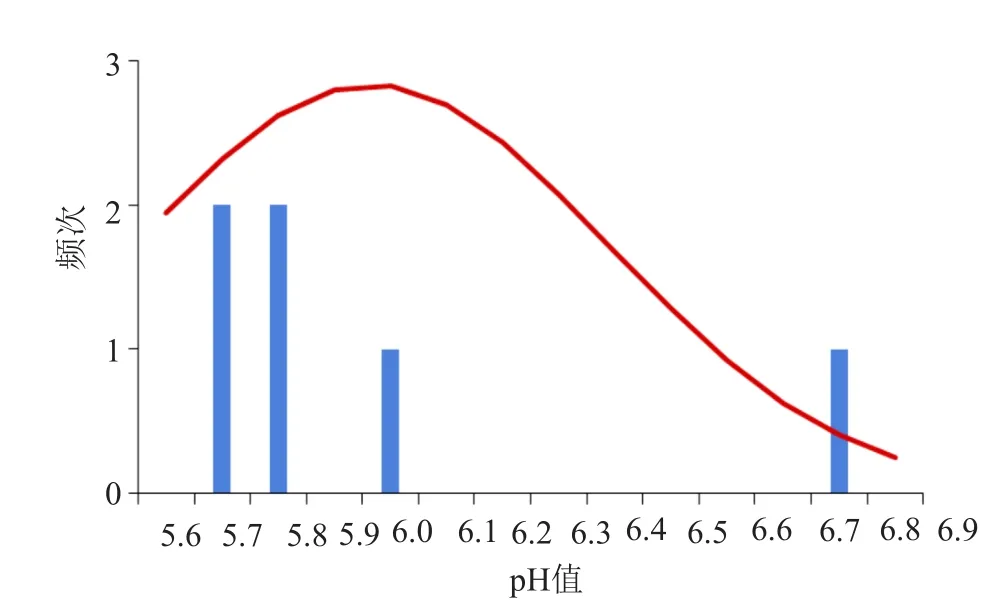
图1 乳糖酸阿奇霉素注射液pH值频次分布图Fig.1 pH distribution of azithromycin lactate injection
新建立的方法与原标准方法相比,灵敏度更高,分离情况更好,检出杂质个数更多,结果更准确。乳糖酸阿奇霉素注射液、原料和注射用粉针制剂测定结果详见表3~5,典型色谱图详见图2。
结果表明,采用HPLC法测定,注射液中共检出8个杂质,远大于按标准检验的1个杂质,杂质之间均达基线分离。未知杂质Ⅲ为单个最大杂质,含量可达3.9%,杂质总量高达6.4%,按照ChP 2015版注射用阿奇霉素有关物质的限度规定,单个最大杂质不得过0.5%,杂质总量不得过2.0%,注射液样品测定结果远超出标准规定上限,合格率为0。此外,杂质J、未知杂质Ⅰ和Ⅲ的含量与出厂时间呈正相关,随出厂时间延长,杂质含量随之上升,均为降解杂质。注射液中杂质含量远高于注射用粉针制剂和原料。
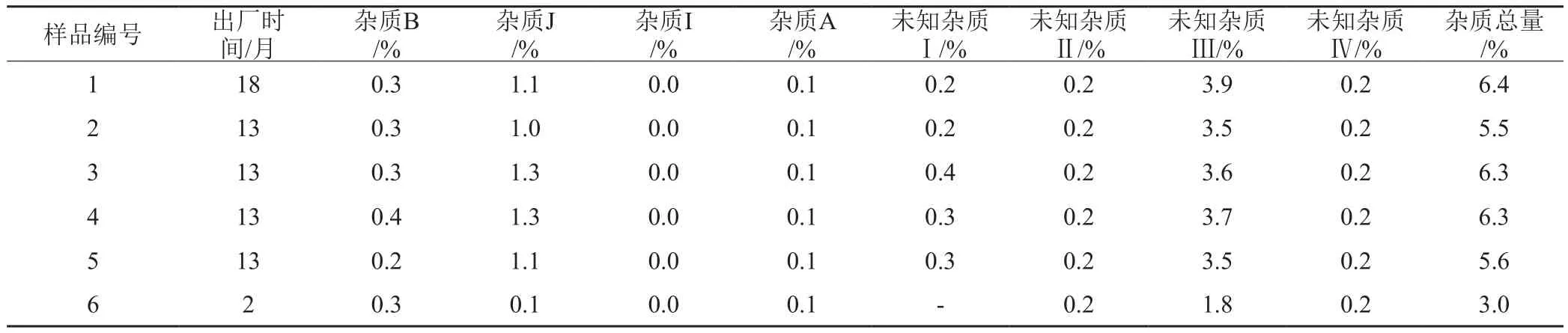
表3 乳糖酸阿奇霉素注射液有关物质结果Tab.3 Results of related substances determination in Azithromycin Lactate Injection
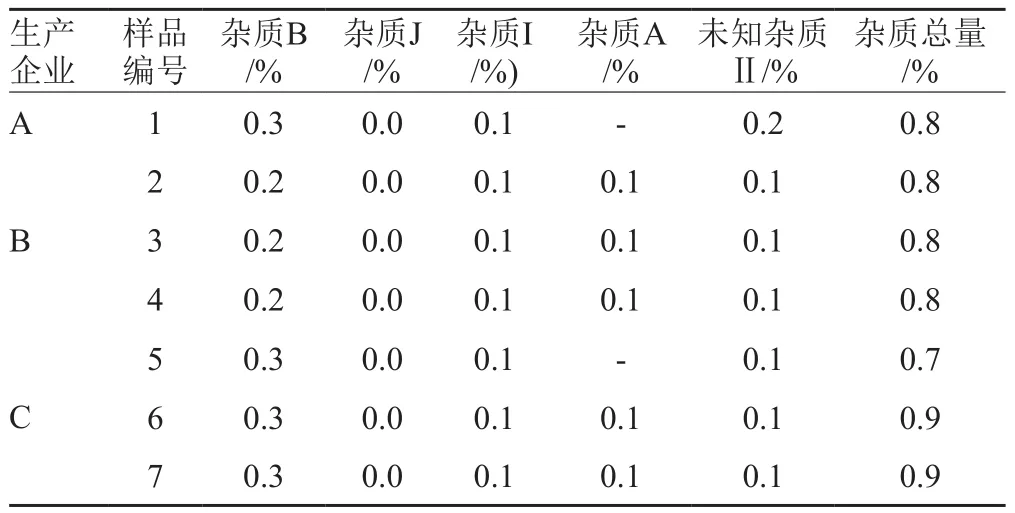
表4 乳糖酸阿奇霉素原料有关物质结果Tab.4 Results of related substances determination in azithromycin lactate
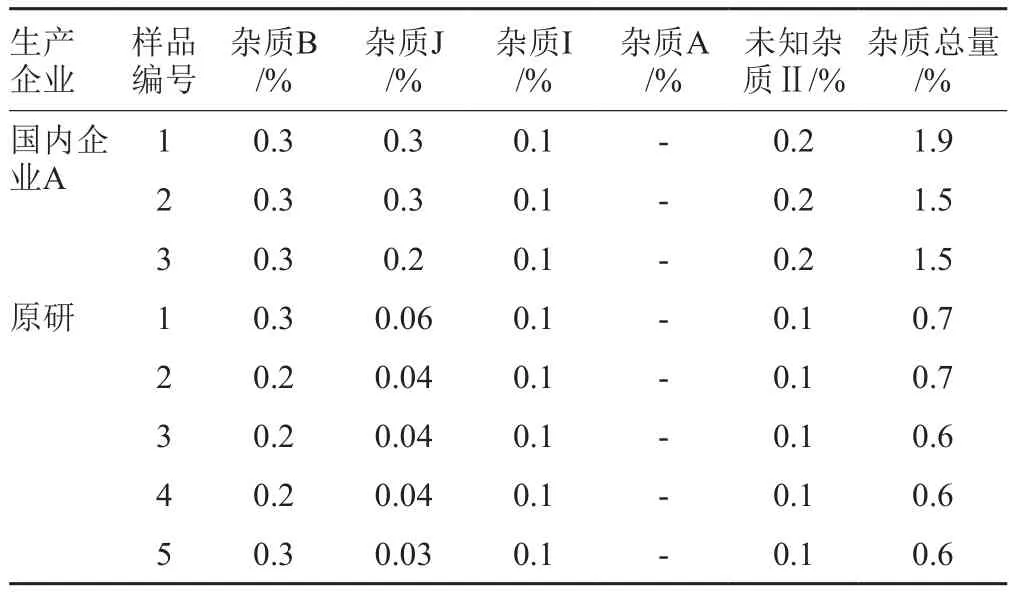
表5 注射用乳糖酸阿奇霉素有关物质结果Tab.5 Results of related substances determination in azithromycin lactate for injection
原研注射用阿奇霉素降解杂质J含量低于0.1%,杂质总量约为0.7%,与乳糖酸阿奇霉素原料中杂质水平相当。国产注射用乳糖酸阿奇霉素杂质总量为1.5%~1.9%,显著高于原研产品。因此,安全性风险也较原研产品高。原料和注射用粉针制剂中杂质主要为来源于阿奇霉素的工艺杂质,杂质含量较低。未知杂质Ⅰ、未知杂质Ⅲ和未知杂质Ⅳ在原料和注射用粉针制剂中均未检出,为注射液剂型特有杂质,且在注射液中含量较高,推测与该注射液剂型相关。
3.2.2 强制降解试验
强制降解试验结果表明,乳糖酸阿奇霉素在固体状态下较稳定,不易降解,在水溶液、室温、偏酸或偏碱性和加热等条件下均降解。在上述条件下,降解产物杂质J和未知杂质III均显著增加。
3.2.3 注射液贮藏条件对杂质影响的考察
由于注射液在常规阴凉处贮藏条件下,降解产物含量较高,因此考察了低温条件贮藏对降解反应进程的影响,结果详见图3。结果表明,低温条件贮藏后降解产物未知杂质Ⅲ含量仍显著增加,而杂质J和未知杂质Ⅰ在出厂时间很短的批次中含量无显著变化,在出厂时间较长的批次中仍显著增加。其余杂质冷藏前后无显著变化。因此,即使低温冷藏也仅能有限的减缓主成分的降解过程,而不能避免降解反应的发生。

图2 乳糖酸阿奇霉素原料和注射剂有关物质典型色谱图Fig.2 Typical chromatograms of related substance in azithromycin lactate and azithromycin lactate injection
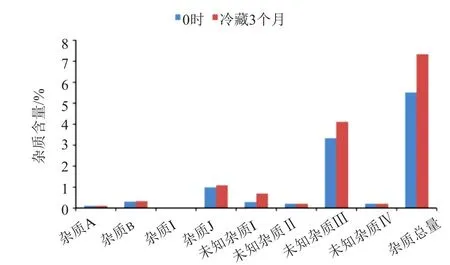
图3 冷藏放置前后有关物质结果比较图Fig.3 Comparison of related substances before and after refrigerated storage
3.2.4 pH值与有关物质相关性分析
溶液贮藏时间对pH值影响:注射液样品冷藏3个月后,与多数出厂时间较长的样品比较,pH值无明显变化;而与一批出厂时间较短的样品(编号6)比较,pH值降低了0.3(表6)。结合该批次样品有关物质的变化,出厂5个月,未知杂质III含量显著增加,pH值显著降低,提示pH值降低可能与未知杂质III密切相关。

表6 冷藏前后乳糖酸阿奇霉素注射液pH值变化Tab.6 pH changes of azithromycin lactate injection before and after refrigerated storage
不同初始pH值对有关物质的影响:不同初始pH值溶液加速试验结果显示,不同温度下放置5d或10d,杂质的变化趋势基本相同,典型的有关物质变化曲线图详见图4。结果表明,未知杂质III随pH升高而增加,溶液pH值大于7.0后,变化幅度明显增大;未知杂质Ⅰ和杂质J随pH下降而增加,溶液pH值小于6.0时,变化幅度明显增大。杂质总量随溶液pH值升高,呈现先下降后上升的趋势。溶液pH值在6.0~7.0之间时,各降解产物的变化相对平缓,在pH值为6.6时,各杂质含量最低。
3.2.5 已知杂质活性研究
综合分析国内外阿奇霉素相关的质量标准及现有杂质对照品情况,对阿奇霉素中已知杂质J、Q、R、A、I、F、S和L的效价进行研究。结果详见表7。结果显示,杂质A、I及S均具有一定的抑菌活性,杂质Q、R、F、J和L在现有实验条件下均无抑菌活性。
3.2.6 未知杂质结构鉴定

图4 加速试验杂质变化曲线图Fig.4 Impurity variation curve of accelerated test

表7 阿奇霉素杂质活性测定结果Tab.7 Result of azithromycin impurity activity measurement
按照ICH(人用药物注册技术要求国际协调会)规定,对其中含量超过0.1%的4种未知杂质采用多级高分辨质谱进行了结构推断,推测的未知杂质I、III和IV结构见图5。推测未知杂质III为阿奇霉素在碱性条件下,内酯环开裂后的降解产物羧酸,因而导致溶液pH降低,与文献中报道的碱水解降解杂质结构一致[13]。未知杂质I和Ⅳ经推测为未知杂质III的成酯化产物。由于其含量在原料及制剂中一致,且与加速反应及贮藏条件均无相关性,仅与配制供试品溶液后放置时间相关,因此推测其来源为主成分阿奇霉素降解后得到未知杂质III,未知杂质III分别与乙醇和甲醇缩合成酯而得。未知杂质Ⅱ经推测为阿奇霉素十五元环上多一个甲基的化合物,为工艺杂质,推测其来源为合成反应最后一步,氮红霉素甲基化后生成阿奇霉素时,发生副反应得到多一个甲基的副反应产物,依现有数据尚不能确定该甲基的具体位置。
3.2.7 含量测定

图5 推测的未知杂质结构Fig.5 The proposed structure of unknown impurities
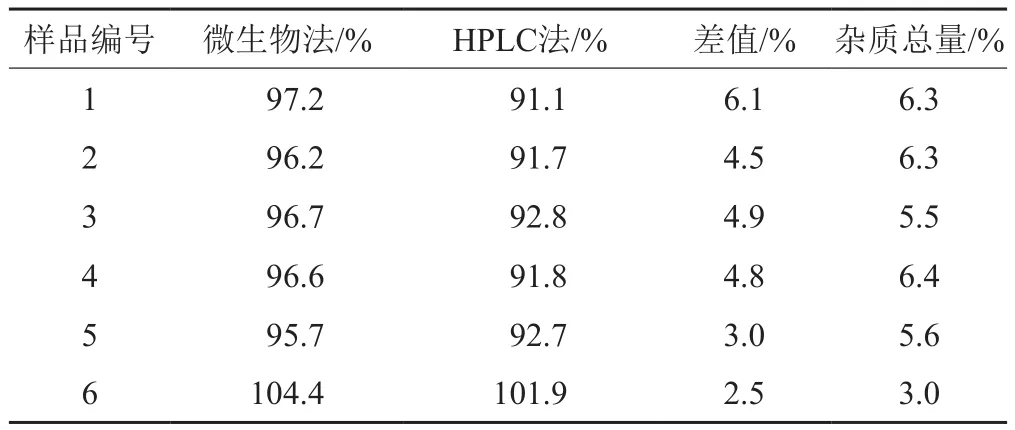
表8 标准方法及拟定方法含量测定结果Tab.8 Content determination results by standard method and proposed method
采用HPLC法测定含量,实现了对供试品中杂质的有效分离,能准确测定主成分含量。注射液样品测定结果详见表8。结果表明,与微生物检定法比较,HPLC法含量测定结果均偏低,两种方法测定结果的差值,随有关物质含量的增加而增大。参考国内外药典及国内注射用阿奇霉素质量标准,现行质量标准的含量限度较宽,建议将含量限度由“90.0%~110.0%”修订为“93.0%~107.0%”。按照ChP2015版注射用阿奇霉素的含量限度“93.0%~107.0%”,6批样品中有5批含量超出低限,不符合规定,合格率为16.7%。样品效期内含量降低明显,与有关物质结果一致,表明该注射液剂型的安全性和有效性均存在较高风险。
4 结论
乳糖酸阿奇霉素注射液现行质量标准中有关物质和含量测定项分别采用薄层色谱法和微生物检定法,专属性差、灵敏度低、操作繁琐,杂质不能有效分离。采用新建立的HPLC法测定有关物质和含量,参照ChP2015版注射用阿奇霉素质量标准,6批次有关物质均不符合规定,5批次含量不符合规定。样品中单个最大杂质可达3.9%,杂质总量可达6.4%,均远超出限度规定,主成分含量偏低,提示产品的质量存在较大风险。
注射液中杂质含量显著高于注射用粉针剂和原料中杂质,现有条件下通过优化低温环境保存和改善溶液pH,仅能有限减缓主成分的降解过程,并不能有效避免杂质含量的升高。同时,原研企业无注射液剂型,国内对注射液剂型未经深入研究即临床应用,为患者健康带来较大风险,剂型合理性为影响本品质量的主要因素。
杂质谱研究表明,注射液中特有杂质未知杂质III为主成分内酯环开裂而生成的羧酸,导致产品pH随出厂时间逐渐降低。溶液pH低于6.0或高于7.0条件下,主成分加速水解,现行质量标准的pH项限度宽泛,需修订pH值限度,并增加有效的pH稳定剂以稳定样品pH值抑制降解。已知杂质的活性研究表明仅少部分杂质具有一定抑菌活性,且活性显著低于阿奇霉素,产品中杂质含量的升高严重影响产品有效性和安全性。
综上分析,建议提升该品种的质量标准,促使企业提升产品质量或淘汰不合理剂型产品。
致谢:本研究工作得到了中国食品药品检定研究院胡昌勤教授及抗生素室诸位老师的大力支持和帮助,谨此致谢。
