灰树花子实体中 水溶性碳水化合物的提取与纯化
2019-03-28,,,,*,,,,,
,, ,,*,,, ,,
(1.江南大学食品学院,江苏无锡 214122; 2.江南大学食品科学与技术国家重点实验室,江苏无锡 214122)
灰树花是一种珍稀的食、药兼用大型真菌,属于多孔菌属,具有强化脾脏[1]、润肺保肝[2]的功效。灰树花富含碳水化合物(含量约为50%)、蛋白质、维生素、矿物质和酚类化合物,脂肪含量较低[3-4]。日本在1982年首次实现了大规模灰树花商业生产[5],至1999年种植面积近4万顷;上世纪90年代,美国和中国也开始了大规模人工培育,2001年中国灰树花种植面积为1.46万顷[6-7]。
灰树花子实体和液体培养菌丝体含有许多活性物质。1984年Ohno等[8]首先指出灰树花多糖具有抗肿瘤活性;1992年美国国家癌症研究院证实灰树花的萃取物有抵抗艾滋病病毒的功效[9];2002年,Kodama等[10]对22~57岁年龄段的肿瘤患者服用灰树花多糖后的疗效进行了调查,结果表明灰树花多糖对不同肿瘤的活性不同。2015年,Xiao等[11]发现,灰树花多糖通过提高胰岛素敏感性,提高磷酸化IR蛋白水平(Try1361)和磷酸化IRS-1水平(Ser307),降低空腹血糖水平。Chen等[12]在体外试验中证明:G. frondosa多糖对DPPH,羟基和超氧化物自由基有清除作用,以及还原能力、亚铁离子螯合作用和抑制大鼠肝脏脂质氧化的能力。但目前对于灰树花中低聚糖的报道较少。
本文进行了灰树花中水溶性碳水化合物的提取、纯化分离和低聚糖的结构鉴定。首先常温(30 ℃)提取可溶性糖(单糖和低聚糖),然后对可溶性糖进行分离纯化和核磁鉴定。对可溶性多糖进行凝胶色谱柱分离并测定了分子量分布。另外,对灰树花原料及提取过程中的结构变化进行了SEM表征,以期为多糖高温提取方面的研究提供参考。
1 材料与方法
1.1 材料与仪器
灰树花子实体 浙江庆元高山农产品批发商部;木瓜蛋白酶 南宁庞博生物工程有限公司;732阳离子交换树脂和D-葡萄糖(AR) 国药集团化学试剂有限公司;海藻糖标准品(纯度99%) 阿拉丁;血糖测定试剂盒 北京利德曼生化技术有限公司;D392弱碱性阴离子交换树脂和711阴离子交换树脂 天津南开和成科技有限公司;SephadexTMG-25、SephadexTMG-75 GE Healthcare Bio-Sciences AB;Dextran系列标品 Sigma公司;Shodex Stangdard P-82标品 Shodex公司。
UV-1100型紫外可见分光光度计 上海美谱达仪器有限公司;DHL-A电脑数显恒流泵 上海沪西分析仪器厂;SBS-100数控记滴自动部分收集器 上海沪西分析仪器厂;LGC-1025 COLUMN HEATER、1525高效液相色谱仪(配2410示差折光检测器和Empower工作站)、UltrahydrogelTMLinear(300 mm×7.8 mmid×2)、Sugar-PakTM-I(300 mm×6.5 mm) 美国Waters公司;SUGAR KS-803 和KS-804(300 mm×8.0 mm) Shodex公司;L-2000高效液相色谱仪 日本Hitachi电子公司;Dionex ICS-5000离子色谱仪 美国Dionex公司;AVANCE Ⅲ 400HZ全数字化核磁共振仪 德国Bruker Biospin公司。SU1510扫描电子显微镜 日本Hitachi公司。
1.2 实验方法
1.2.1 灰树花水溶性碳水化合物的提取 灰树花子实体粉碎过80目筛,称取50 g粉末于夹套反应器中,按一定料液比加入去离子水,添加一定量木瓜蛋白酶,先于50 ℃酶解反应1 h,100 ℃灭酶10 min,再在一定温度下机械搅拌一定时间后,5000 r/min离心15 min,得上清液Ⅰ。渣相Ⅰ中加入300 mL去离子水,在高压反应釜中130 ℃下反应4 h,之后5000 r/min离心15 min,去离子水清洗渣相三次,合并上清液,抽滤2次,滤液中加入4倍体积的无水乙醇于4 ℃静置12 h,5000 r/min离心15 min,无水乙醇清洗渣相,待乙醇挥发干净,冻干(0.04 Mbar,-50 ℃)得粗多糖。粗多糖得率(X%)根据式(1)计算:
式(1)
式中,m1为冻干所得粗多糖质量(g);m2为原料子实体干重(g)。
1.2.2 可溶性糖提取纯化及结构鉴定
1.2.2.1 可溶性糖提取的单因素实验 分别考察料液比(w/v,1∶15、1∶20和1∶30 g/mL)、木瓜蛋白酶的添加量(0、0.2%和0.4%)、温度(30、60、70和80 ℃)和反应时间(1、2、3和4 h)4个因素作单因素实验,考察各个单因素对可溶性糖得率的影响。将其中一个作为单因素时,其他三个因素的固定实验水平分别是料液比1∶20 g/mL、温度30 ℃、反应时间2 h和加酶量0.2%。采用苯酚硫酸法[13]测定总糖含量,以总糖含量大小反映可溶性糖的得率;用无水葡萄糖制作标准曲线,回归方程为y=11.352x+0.0055(R2=0.9993)。可溶性糖得率根据式(2)计算:
可溶性糖得率(%)=[总糖浓度(mg/mL)×上清液Ⅰ体积(mL)]/子实体干重(mg)×100
式(2)
1.2.2.2 分子量表征 采用高效凝胶过滤色谱法(HPGFC)测量浸提液中碳水化合物的分子量分布。将30、60、80和130 ℃不同温度提取得到的浸提液,10000 r/min离心10 min,0.45 μm过滤器过滤后进样。色谱条件:色谱柱UltrahydrogelTMLinear(300 mm×7.8 mm);流动相0.05 mol/L NaNO3;流速0.9 mL/min;柱温30 ℃。标准曲线方程:y=13.6-0.559x(R2=0.9974)(其中x为保留时间,min;y为lg(Mp))。
1.2.2.3 可溶性糖纯化 取150 mL水酶法提取得到的上清液Ⅰ,向其中加入15 g 732树脂,20 ℃下200 r/min静态吸附30 min后抽滤,取滤液,向其中添加15 g 711树脂,20 ℃下200 r/min静态吸附30 min后抽滤,取滤液,再向其中添加2 g 732树脂和4 g 711树脂,20 ℃下200 r/min静态吸附15 min脱除残余盐分。最后向滤液中加入20 g D392树脂,在60 ℃下200 r/min脱色2 h得到无色澄清溶液,命名为纯化液。将纯化液浓缩至原体积的1/4,得到浓缩液。分别测定上清液Ⅰ、纯化液和浓缩液的固形物含量、含盐量、色素和蛋白质含量(其中,用分光光度计A420处的吸光值表示色素含量[14];凯氏定氮法测定蛋白含量[15],换算系数4.38[16];阿贝折光仪测定固形物含量;电导率仪测定溶液电导率,用以表示溶液中盐的情况[17])。根据式(3)和(4)分别计算脱色率(η)和蛋白质质量浓度(c2):
式(3)
式中,A0和A1分别是上清液Ⅰ脱色前和脱色后在420 nm下的吸光度。
式(4)
式中,c1和v1分别为盐酸的浓度(mol/L)和体积(mL);f为换算系数;v2试样体积。
1.2.2.4 可溶性糖的凝胶色谱层析分离 将浓缩液(总糖浓度为48.25 mg/mL)10000 r/min离心15 min,0.22 μm滤膜过滤后,用Sephadex G-25凝胶柱(10 mm×100 cm,分离范围100~5000 Da)进行分离。柱层析条件:空水体积32 mL;上样量2 mL;纯水洗脱;洗脱速度0.1 mL/min;定滴收集2 mL/管。
1.2.2.5 HPLC分析 合并洗脱曲线相同组分进行HPLC分析。根据可溶性糖分子量分布结果,选择水苏糖、海藻糖、木糖、半乳糖、果糖和葡萄糖为标品。样品经10000 r/min离心10 min,0.45 μm滤膜过滤后进样。色谱条件:色谱柱Sugar-PakTM-I(6.5 mm×300 mm);去离子水为流动相;柱温85 ℃;流速0.40 mL/min;进样体积10 μL;示差折光检测器。
1.2.2.6 可溶性糖中低聚糖的NMR分析 将凝胶层析收集到的单一样品冻干(0.04 Mbar,-50 ℃),配制成浓度30 mg/mL的待测样,进行13C和1H分析。参照物为海藻糖标品(30 mg/mL)。核磁条件:磁场强度9.4 Tesla,碳谱的射频频率100 Hz,氢谱的射频频率400 Hz,5 mm Z梯度场正向多核探头
1.2.2.7 可溶性糖中海藻糖含量测定 称取2.0000 g海藻糖标准品,配制成20 mg/mL标准液。依次用水稀释成15、10、5、2、1、0.5、0.25 mg/mL的溶液。上清液Ⅰ10000 r/min离心10 min,0.45 μm过滤器过滤后进样,HPLC条件同1.2.2.5。以海藻糖的峰面积为横坐标,浓度为纵坐标,绘制标准曲线,标准曲线的回归方程为y=3×10-6x-0.079(R2=0.9993)。海藻糖含量(ω%)根据式(4)计算:
式(5)
式中,ρ是HPLC测得的海藻糖浓度(mg/mL);V为上清液Ⅰ体积(mL);f为试样稀释倍数;m为试样质量(g)。
1.2.3 灰树花多糖分离纯化及分子量表征
1.2.3.1 粗多糖层析分离及纯度鉴定 多糖冻干样品配制成10 mg/mL的溶液,10000 r/min离心15 min后,上清液经0.45 μm滤膜过滤。用Sephadex G-75凝胶(16 mm×100 cm,分离范围1~50 kDa)分离,柱层析条件:空水体积53 mL;上样量2 mL;纯水洗脱;洗脱速度0.24 mL/min;80滴/管。苯酚-硫酸法测定总糖含量,绘制洗脱曲线。合并相同组分,HPLC分子量表征和鉴定纯度,色谱条件:色谱柱Shodex SUGAR KS-803、KS-804;柱温60 ℃;流动相0.1 mol/L NaNO3溶液;流速0.6 mL/min;检测器示差折光检测器。标准曲线为 y=9.6717-0.2156x(R2=0.9967)(其中x为保留时间,min;y为lg(Mp))。
1.2.3.2 SEM表征 取少量灰树花子实体原料粉末和冻干的渣相Ⅱ粉末(130 ℃提取离心后所得)均匀粘在导电胶上,喷金制样,操作电压为5 kV,观察其微观结构。
1.3 数据处理
2 结果与分析
2.1 可溶性糖提取、纯化及结构鉴定结果
2.1.1 可溶性糖提取的单因素实验结果 图1(a)表明,温度为30、60、70和80 ℃时得率无显著性差异(p>0.05),说明温度对得率影响不大,因此采用30 ℃进行常温提取,可溶性糖得率为21.48%,图1(b)表明,添加少量木瓜蛋白酶可以明显提高可溶性糖得率(p<0.05),这是因为木瓜蛋白酶能够切断糖与蛋白的连接,使糖容易浸出。而加酶量在0.2%与0.4%时,低聚糖得率无显著性差异。图1(c)表明,可溶性糖得率在料液比1∶15 g/mL时,明显低于料液比1∶20和1∶30 g/mL的得率(p<0.05),且当料液比高于1∶20 g/mL时可溶性糖得率无显著性差异(p>0.05),因此可确定料液比为1∶20 g/mL。图1(d)表明,提取时间2、3和4 h时,可溶性糖得率无显著性差异(p>0.05),因此确定提取时间为1 h。通过单因素实验确定总糖的提取条件为:温度30 ℃;木瓜蛋白酶添加量0.2%;料液比1∶20 g/mL;提取时间1 h,结果表明,该工艺条件下,可溶性糖得率为23.87%。
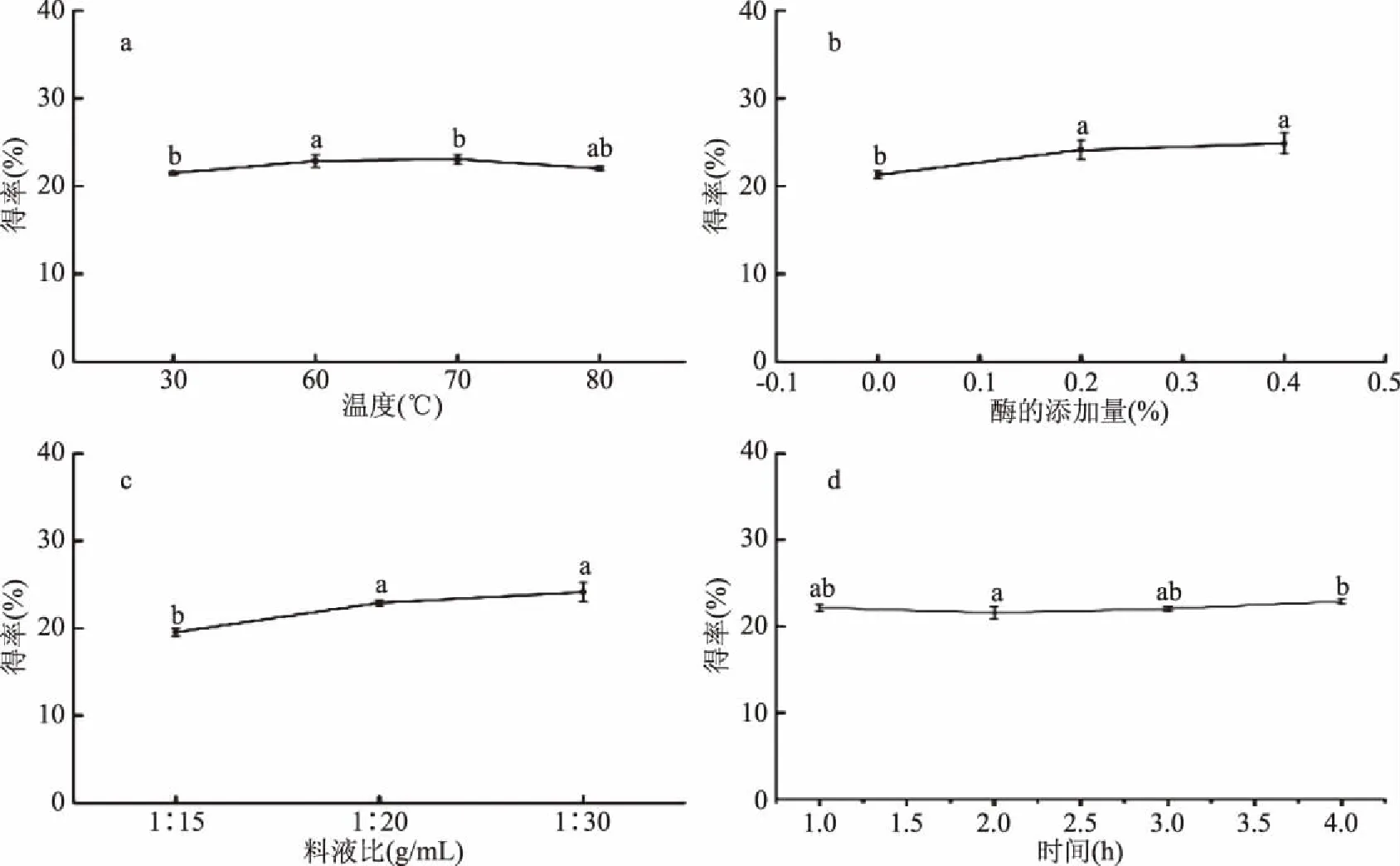
图1 不同提取条件对得率的影响Fig.1 Effect of different extraction conditions on the extraction yield
2.1.2 分子量表征 30、60和80 ℃所提取的可溶性糖分子量分布结果如图2所示。结果显示可溶性糖的主要信号出现在260 Da左右,考虑到分子量分布检测的误差,该信号可能是单糖(180 Da)或二糖(342 Da),需要进一步验证;在19 min附近的信号分子量为1 kDa左右,可能是聚合度为6~7的寡糖;
在17 min附近的微弱信号对应于分子量15 kDa左右的多糖,推测其聚合度为90左右,可能是更大分子量多糖降解产生的碎片。随着提取温度升高,发现所得到的可溶性糖中的多糖片段含量有所升高,在图2(c)中还发现了很少量分子量在760 kDa左右的多糖成分。理论上,提高温度使分子运动加剧,使得一些在常温下不溶的多糖变的可溶,但由于多糖分子量大且分子之间形成的氢键数量较多,80 ℃还不足以完全打破多糖之间的强分子相互作用而导致多糖溶出。图2(d)为130 ℃下提取的可溶性糖分子量分布,可以发现高温浸提时多糖的溶出程度明显提高,包括较多分子量约为300 kDa的多糖和一些分子量约14 kDa的多糖片段,甚至出现了少量2×105kDa的高分子量多糖。

图2 不同温度浸提液中碳水化合物的分子量分布Fig.2 The Molecular weight distribution of carbohydrates in different temperature extracts注:a:30 ℃;b:60 ℃;c:80 ℃;d: 130℃。
2.1.3 可溶性糖纯化 根据以上研究结果,制备了30 ℃常温提取的上清液I,并进一步进行纯化以表征鉴定其中的可溶性糖。经树脂脱色脱盐后,上清液I中的色素、盐分、蛋白和总糖的情况如表1所示,纯化液的电导率只有119 μs/cm,根据式(3)计算得脱色率为89.43%。由于蛋白质为带电荷的两性物质,脱盐脱色过程中,蛋白质可吸附于弱碱及弱酸性树脂,从而被有效脱除,最终蛋白质脱除率为93.65%。糖是一种多羟基化合物,所用离子交换树脂也能吸附糖类,最终纯化过程中糖的损失率为34.19%。

表1 上清液Ⅰ、纯化液和浓缩液色素、蛋白、总糖和电导率情况Table 1 The content of pigment,protein,total sugar and conductivity in SupernatantⅠ,Purification and concentrate solution
2.1.4 可溶性糖的凝胶色谱层析分离 纯化后的上清液I经浓缩后在G-25凝胶柱上进行层析分离,结果如图3所示,得到两种组分,流出体积分别位于38 mL和75 mL,峰形对称且分离较好。因此选择了其中6管(第19、35、36、37、38和39管)进行HPLC分析。HPLC所用各种糖标准品的保留时间(min)如表2所示。HPLC结果见图4,如图4(a)所示,第19管样品含量低且至少包含了3个组分,可能是相对分子量较大的寡糖或多糖片段,即对应于图2(a)结果。第35、36和37管(图4(b)~(d))均为单一样品,信号位置根据标准品判断为海藻糖(RT=9.38 min),且从35至37号管海藻糖含量逐渐升高。第38号管(图4(e))对应于G-25层析结果的峰下行位置,HPLC结果显示其除了含有海藻糖以外,还含有葡萄糖(RT=11.95 min),另外在16 min处还有一个小信号,可能是另一种含量很少的单糖或残余杂质。而在第39号管(图4(f))中可见海藻糖含量已经低于葡萄糖,同时两种糖的含量均明显下降。因此可以推断,在图2(a)中250 Da处的信号应该包含了海藻糖和葡萄糖,而它们在G-25层析柱中也没有得到很好地分离,都包含在流出体积65~83 mL范围的尖峰之中。进一步地,根据表2可初步判断上清液Ⅰ中的低聚糖为海藻糖。

表2 标准品的液相出峰时间Table 2 Peak time for standard products

图4 不同收集管的液相图Fig.4 The liquid phase diagram of different collecting tube注:a:管19;b:管35;c:管36;d:管37;e:管38;f:管39。

图3 G-25的柱层析图Fig.3 G-25 Column chromatogram
2.1.5 NMR结构鉴定 为确证HPLC中9.38 min处物质为海藻糖,进一步采用NMR鉴定样品结构。结果如图5和表3所示,该化合物的各个H原子和C原子的化学位移与海藻糖的标准品基本一致,且质子数、碳原子数与海藻糖的分子式一致。其中,端基质子δ5.20(d,J=3.8 Hz)为耦合常数3.8 Hz的二重峰,属于D-葡萄糖的1位质子与2位质子呈e-α立体关系,表明为α立体构型。二糖异头碳位的化学位移完全一致,表明二糖分子结构对称。综上,可以确定样品为(α,α)-海藻糖。
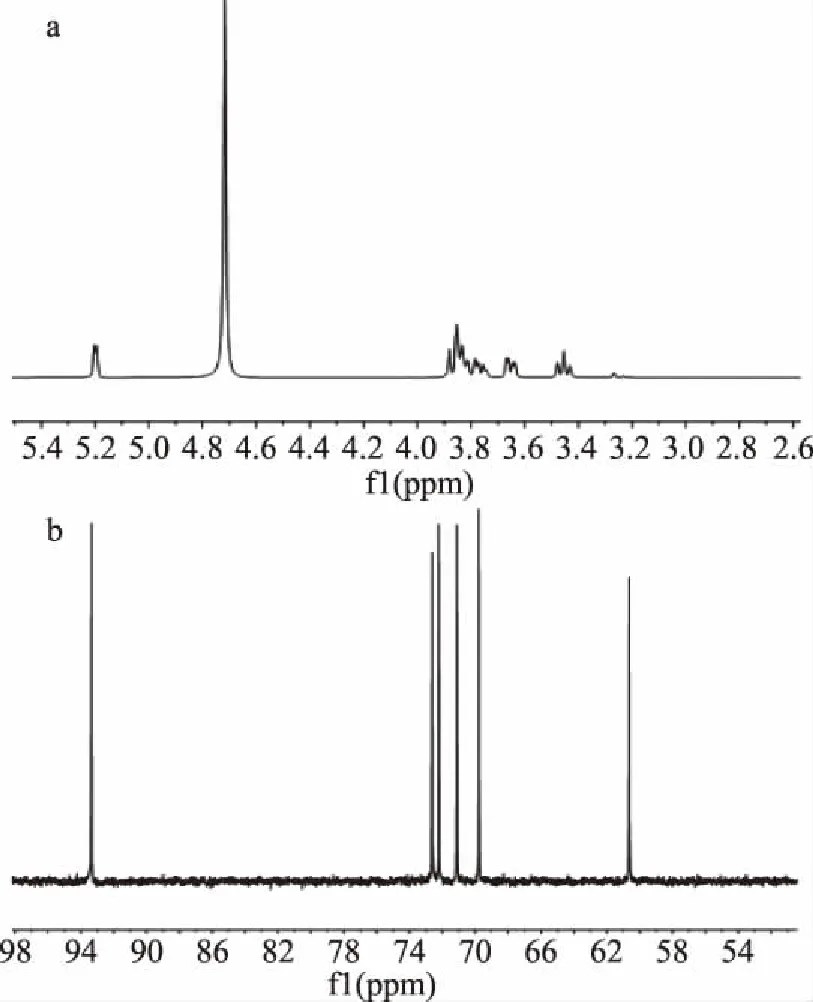
图5 样品的13C(a)和1H(b)图Fig.5 13C(a)and 1H(b)spectrum of sample

δC海藻糖标品/样品δH海藻糖标品/样品93.32/93.335.20/5.20,d,J=3.8 Hz72.62/72.623.84/3.85,m72.23/72.233.76/3.76(dd),J=11.8,5.1 Hz,1H71.12/71.123.65/3.65,dd,J=9.9,3.8 Hz69.79/69.793.45/3.45,t,J=9.4 Hz60.64/60.63-
2.1.6 可溶性糖中海藻糖的定量 上清液Ⅰ经纯化、分级、HPLC分析和NMR鉴定后,证实了上清液Ⅰ中的低聚糖为海藻糖。HPLC定量分析的结果显示:上清液Ⅰ中海藻糖的峰面积为1133368,根据式(5)算得灰树花中海藻糖的含量为10.98%(占干重)。
2.2 灰树花多糖分离纯化及分子量表征结果
2.2.1 多糖的提取得率 多糖提取的基本依据是多糖的结构和溶解性。多糖结构复杂,因技术限制无法分析透彻,而且灰树花多糖的相对分子质量大,水溶解性低,大多通过氢键、离子键等与其他多糖等物质聚合在一起,须要通过有效的方法破坏结合键才能实现多糖的提取。故利用高温破坏多糖与其他物质的结合键并提高多糖的溶解度,以增加多糖的提取率。实验结果表明:经高温水提、醇沉得到的多糖得率为3.42%。
2.2.2 多糖的纯化 图6表4表明,G-75柱层析图显示的第一个高峰主要含有两种多糖组分,其分子量分别为300 kDa和16 kDa。收集管23~27是分子量为318 kDa左右的多糖,收集管28~38是分子量为300 kDa和16 kDa的混合物,而且随着流出体积的增大,分子量为16 kDa的多糖成分增多,符合凝胶分离的尺寸排阻原理。流出体积在180 mL附近的小峰,因多糖含量太低,所以无法用示差检测器检测到信号,根据流出时间推测其分子量为1 kDa左右的多糖组分。

图6 G-75的柱层析图Fig.6 G-75 Column chromatogram

收集管号峰的编号保留时间(min)分子量(kDa)相对峰面积(%)23~27119.3331810028~30119.4530088.87225.351611.1331~33119.4030858.99225.391541.0134~38119.1035720.13225.351679.87
2.2.3 扫描电子显微镜 图7(a)为灰树花子实体原料颗粒的显微观察照片,其表面为比较致密且粗糙的形态;图7(b)为原料放大观察照片,可见子实体为层状堆积结构,未见孔道结构。图7(c)和7(d)为高温提取后灰树花渣相Ⅱ的观察图,呈现多孔网状结构。根据Michalenko等[18]报道,食用菌的细胞壁主要成分是几丁质、β-葡聚糖和蛋白质,细胞壁外层主要是用于抵抗外界压力水溶性葡聚糖;内层是作为细胞壁骨架的水不溶性葡聚糖和几丁质。灰树花中的海藻糖、葡萄糖以及部分多糖溶解后,其细胞壁结构遭到一定破坏,内部骨架暴露,整体结构呈现疏松多孔状态。

图7 电子显微镜图Fig.7 Electron microscopes注:原料(a):×50;原料(b):×500;水提后渣相Ⅱ(c):×50;水提后渣相(d):×500。
3 结论
灰树花的常温浸提液经脱色、脱盐和脱蛋白后,再通过凝胶过滤处理得到单一低聚糖,高效液相和核磁共振实验得出结论:灰树花中的低聚糖为海藻糖,含量约为10%。经高温处理,多糖的提取率为3.42%,凝胶过滤得到两种单一组分的多糖,高效液相鉴定其分子量分别为300 kDa和16 kDa。本研究对灰树花低聚糖和多糖探索为后期深入探索灰树花碳水化合物的理化性质和功能做了初步准备。
