二氧化锆QuEChERS-高效液相 色谱-串联质谱测定鱼肉中 2种硝基咪唑及代谢产物
2019-03-28
(福建省食品药品质量检验研究院,福建福州 350000)
硝基咪唑类药物是一类5位被硝基取代的杂环化合物,主要包括甲硝唑、地美硝唑、洛硝哒唑、替硝唑等[1]。该类药物有抗菌和抗原虫作用,并具有成本低、疗效快的特点,被广泛应用于水产品养殖中寄生虫的防治与感染。研究表明,该类药物及其代谢产物具有潜在的致癌、致病变及损伤DNA的作用[2],因此,我国对于硝基咪唑类药物在动物源性食品养殖及生产过程中的应用进行了严格的控制[3-4]。
目前,测定硝基咪唑类抗生素及其代谢物的方法主要有气相色谱法[5]、气相色谱质谱联用法[6]、高效液相色谱法[7]和高效液相色谱质谱联用法[8]等。由于液相色谱法的检出限较高,基质干扰较大,难于定量;气相色谱法及气相色谱质谱联用法抗干扰能力较弱,适用范围相对较窄;而液相色谱质谱联用法同时具有较低的检出限、较高的抗干扰能力及适用范围广等特点,被广泛应用于硝基咪唑及其代谢产物的检测。我国现行有效的动物源性食品及饲料[9]中硝基咪唑及其代谢物检测标准中检测方法主要是液相色谱串联质谱法,故而本文选取该方法为检测方法。
硝基咪唑及其代谢产物残留检测常用到的前处理方法有液液萃取[10]、固相萃取[11]和QuEChERS[12]等。QuEChERS技术因高效、快速、成本低等特点,被广泛地应用于农兽药残留检测领域[13]。常规用于兽药检测前处理的QuEChERS试剂成分主要是十八烷基键合硅胶吸附剂、N-丙基乙二胺吸附剂和无水硫酸镁,可满足大部分动物源性食品中兽药残留的检测,但是对于磷脂含量较高的食品,经QuEChERS试剂包处理后,仍会出现基质干扰,导致灵敏度和测量准确性降低。锆离子或锆氧化物与磷酸根或基团具有强的配位作用,Xiangyang等[14]利用UIO-67中的锆氧簇中的锆来吸附水样中的草甘膦和草胺磷,彭西甜等[15]、姚凯等[16]利用该机理用纳米级的二氧化锆吸附脂类及磷脂。本文同样基于该机理,在QuEChERS试剂中加入二氧化锆颗粒用于吸附磷脂来降低基质干扰,同时参考已有的研究和标准,拟建立改进的QuEChERS方法结合HPLC-MS/MS来测定动物源性食品中的硝基咪唑类药物,并以鱼肉为代表性基质,以两种硝基咪唑药物和一种代谢产物为目标兽药,并对实验条件及QuEChERS试剂量进行优化,以期满足鱼肉中硝基咪唑及其代谢产物残留的快速检测。
1 材料与方法
1.1 材料与仪器
甲硝唑(Metronidazole,MNZ) 中国食品药品检定研究院;羟甲基甲硝咪唑(2-hydroxymethyl-1-methyl-5-nitroimidazole,HMMNI) 德国WITEGA公司;地美硝唑(Dimetridazole,DMZ) Dr. Ehrenstorfer公司;乙酸乙酯、乙腈色谱纯 美国TEDIA公司;乙酸铵、氨水、甲酸 色谱纯,上海阿拉丁公司;二氧化锆(粒径200~400 nm) 美国Sigma公司;十八烷基键合硅胶吸附剂(C18)、N-丙基乙二胺吸附剂(PSA)、无水硫酸镁 博纳艾洁尔公司;超纯水 由Milli Q系统制得,美国Milipore公司;实验所用的鱼肉 均为市售的鲜活海鲈鱼,宰杀后取可食用部分捣碎所得。
API5500型四极杆串联质谱仪配电子喷雾离子源 美国AB公司;Agilent 1290型超高效液相色谱仪 美国Agilent公司;Agilent Eclipse Plus C18色谱柱(2.1 mm×50 mm,1.8 μm) 美国Agilent公司;Hei-VAP Precision减压旋转蒸发仪 德国Heidolph公司;CR21N高速冷冻离心机 日本HITACHI公司;SW22恒温水浴振荡器 丹麦FOSS公司;VORTEX 4 digital涡旋混合器 德国IKA公司;HM100刀式研磨仪 北京格瑞德曼公司。
1.2 实验方法
1.2.1 标准工作溶液和缓冲液的配制 准确称取各硝基咪唑标准物质10.0 mg,用甲醇溶解并定容至10.0 mL,配得浓度为1.0 mg/mL的单标溶液;准确吸取上述各单标溶液适当体积于100.0 mL容量瓶中,用甲醇定容配得浓度为1000 ng/mL的三种物质混标标准中间溶液;于4 ℃下避光保存。称取乙酸铵14.4 g用9000 mL水溶解,再用氢氧化钠溶液调节pH至5.2,然后定容至1000.0 mL混匀后得乙酸缓冲液。
1.2.2 液相色谱与质谱条件 Agilent Eclipse Pluse C18色谱柱(2.1 mm×50 mm,1.8 μm)。流动相:0.1%甲酸水(A)-0.1%甲酸乙腈(B);梯度洗脱程序:0~3.0 min,95%~5% A;3.0~5.0 min,5%~5%;5.0~5.1 min,5%~95%;5.1~7.0 min,95%~95%;柱温:30 ℃;流速:0.25 mL/min;进样量:1.0 μL。本实验着重考察了0.1%甲酸水-甲醇、0.1%甲酸水-乙腈、0.1%甲酸水-0.1%甲酸甲醇、0.1%甲酸水-0.1%甲酸乙腈做为流动相时,对各物质的峰型及分离效果的影响。
电子喷雾离子源(ESI),正离子扫描,多反应监测扫描模式(MRM);离子源温度:450 ℃;电喷雾电压:4500 V;气帘气压:30 Psi;雾化气压:55.0 Psi;辅助气压力:55.0 Psi;碰撞室入口电压:10 V;碰撞室出口电压:15 V。
在优化质谱条件时,采用直接进样的方式,分别将50 μg/L的单标液注入离子源,在ESI正离子模式下,获得准分子离子峰。参考欧盟2002/657/EC的规定,以准分子离子峰为母离子,选择响应值较高的两个碎片作为子离子,和母离子组成离子对,同时分别优化各离子对的碰撞能量、去簇电压、碰撞室出口及入口电压。为保证三种硝基咪唑的检测灵敏度和准确度,本文采用多反应监控模式(MRM),见表1。

表1 3种硝基咪唑类的多反应监测扫描模式下的质谱参数Table 1 Mass spectrometric conditions (MRM)of three nitromidazoles
1.2.3 样品前处理方法及优化 鱼肉样品的制备:取鱼的可食部分,刀式研磨仪以8000 r/min 的速度,2 min充分捣碎混合均匀后,分装入洁净的聚四氟乙烯材质的小瓶中,密封,并于-18 ℃以下冷冻存放,每次使用前取一份分装样品,常温充分解冻,待回温至室温后称取,且每份分装样品解冻称取后即弃去。
称取2.00 g鱼肉于50 mL离心管中,加入8 mL乙酸铵缓冲液,涡旋混匀,室温静置,加入15 mL提取溶剂、5.0 g无水硫酸钠,高速涡旋混匀后,放于室温水平振荡器中,振荡提取10 min,在4 ℃条件下,以9500 r/min的高速离心5 min,取上清液;残渣重复提取一次后,合并两次提取液,待净化。向提取液中加入净化剂(100 mg PSA、40 mg C18、600 mg无水硫酸镁及一定量的二氧化锆),高速涡旋1 min混匀,在4 ℃条件下,以9500 r/min的高速离心5 min,吸取全部有机相,40 ℃下在减压旋转蒸发仪上蒸发至近干,精密加入1 mL 0.1%甲酸水-乙腈(95∶5,体积比),涡旋溶解残渣,过0.22 μm滤膜,待上机。
1.2.3.1 提取溶剂的选择 按照1.2.2仪器条件及1.2.3提取过程,考察不同提取溶剂对样品中硝基咪唑的提取效率,本文比较1%氨水-乙腈、1%氨水-甲醇、1%氨水-乙酸乙酯的提取效率,在其他实验条件不变的前提下,提取效率是通过加入相同量(3 μg/kg)标准溶液,不同提取溶剂的回收率高低来确定。
1.2.3.2 二氧化锆加入量的确定 考察不同加入量的二氧化锆,对样品基质净化效果的影响。在本文中,C18、PSA和无水硫酸镁的量参照SN/T 3235-2012中的量[17]加入,对二氧化锆的量(0、10、20、40、60、80、100 mg)进行优化。通过比较在加入相同加标量时(3 μg/kg),三种硝基咪唑峰面积的大小,确定最佳的二氧化锆加入量。
1.2.4 基质效应计算 基质效应是质谱检测过程中影响方法灵敏度、精密度及准确度的主要因素之一[18]。本文采用相对响应值法来评价三种硝基咪唑类的基质效应:
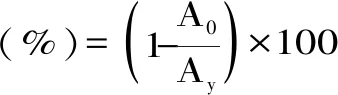
式(1)
式中:Ay为基质配标响应值,A0为溶剂配标响应值。当基质效应的为正值时,表现为基质增强效应;当基质效应为负值时,表现为基质抑制效应;基质效应的绝对值越大,基质效应越强。通常认为,基质效应的绝对值小于20%时,可以认为基质干扰对结果的影响不大,但当其绝对值大于20%时,基质效应不可忽略。
1.2.5 标准曲线、检出限及定量限 按照1.2.3步骤处理空白样品得空白基质溶液,取适量的标准中间溶液,以空白基质溶液定容至质量溶度分别为0.5、1.0、2.0、5.0、10.0、20.0 ng/mL的标准工作溶液。按照1.2.2的条件进样,以质量溶度为横坐标,以分析物的峰面积为纵坐标,绘制标准曲线。方法的检出限(LOD)和定量限(LOQ)是通过空白样品加入不同浓度的标准工作溶液的方法测定的,其中当信噪比RS/N≥3.0时的含量为检出限,RS/N≥10.0时的含量为定量限。
1.2.6 回收率及精密度实验 取空白鱼肉样品,三种硝基咪唑的添加浓度依次为:MNZ 0.2、0.4、1.0 μg/kg,DMZ 0.2、0.4、1.0 μg/kg,HMMNI 0.5、1.0、2.5μg/kg三个水平标液,每个浓度平行6次试验,前处理方法参照1.2.3,仪器条件参照1.2.2,进行检测,计算回收率和相对标准偏差。
1.3 数据处理
采用Analyst及MultiQuantTM软件进行分析,采用Origin 8.0作图。
2 结果与分析
2.1 色谱及质谱条件的优化
在正离子模式条件下,甲酸的加入可以增大离子化效率[17],同时参考了文献及现有的国家标准,甲酸的含量在0.1%时,结果最优。所以本实验着重考察了0.1%甲酸水-甲醇、0.1%甲酸水-乙腈、0.1%甲酸水-0.1%甲酸甲醇、0.1%甲酸水-0.1%甲酸乙腈做为流动相时各物质的分离效果。对比各流动相的标液谱图,发现以0.1%甲酸水-0.1%甲酸乙腈作为流动相时,分离效果最佳,峰型较好无拖尾现象,故在本文中选用0.1%甲酸水-0.1%甲酸乙腈作为流动相,三种化合物的总离子流图见图1。
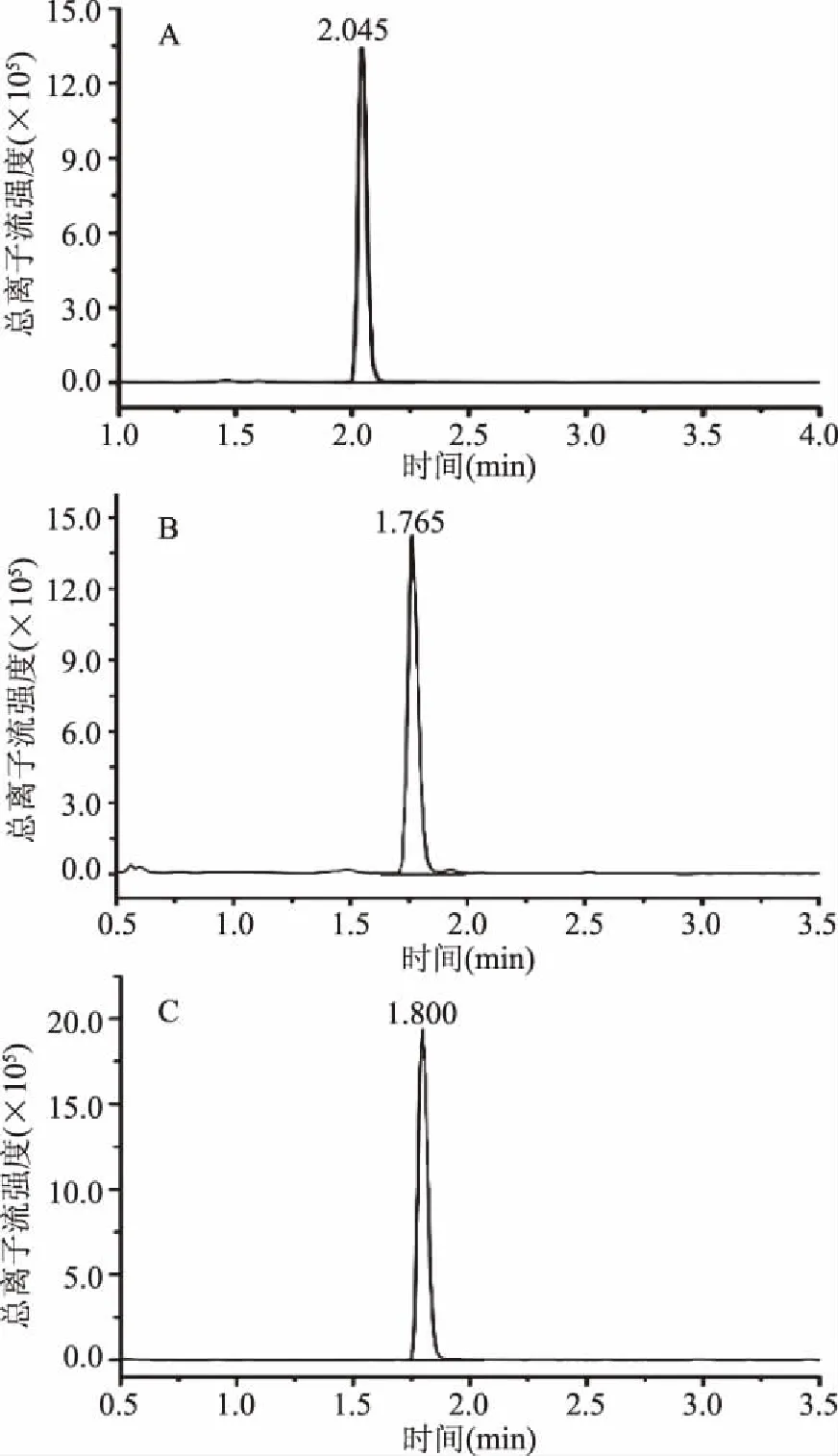
图1 3种硝基咪唑类药物的定量离子流图Fig.1 MRM chromatograms of quantitative ion pairs for 3 nitromidazoles注:A、B、C依次为DMZ、HMMNI、MNZ。
2.2 提取溶剂的优化
在检测硝基咪唑类药物时,常被用到的提取剂主要有乙腈[12,19-21]、乙酸乙酯[22]、甲醇[23]及一些混合试剂。硝基咪唑类药物是酸碱两性化合物,在弱酸性条件下可以质子化,在弱碱性条件下则以游离分子形式存在,据文献[12]报道,DMZ、MNZ两种对提取溶剂的pH要求不高,但是HMMNI的提取效率在中性或者酸性条件下较低,故本文在提取剂中加入了氨水。如图2所示,1%氨水-甲醇的提取效率最低;1%氨水-乙酸乙酯的提取效率最高,且在旋蒸过程中所需的温度低,时间短;1%氨水-乙腈提取效率相对较高,同时乙腈可以有效地沉淀动物源性食品中的蛋白质,且脂肪提取率低,故而基质影响最小。综合考虑后,最后选择1%氨水-乙腈作为提取剂。
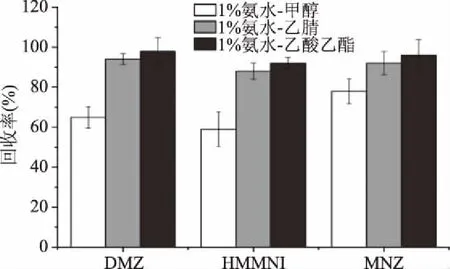
图2 提取剂对硝基咪唑类药物提取回收率的影响Fig.2 Effects of different solvent combination on the recovery rate of three nitromidazoles
2.3 二氧化锆加入量的优化
在兽药残留量分析的过程中,分散固相萃取法常用到的净化吸附剂有C18、PSA和无水硫酸镁三种,可以高效地去除样液中的脂肪、色素、金属离子和有机酸等,但是动物源性产品中磷脂含量较高,且磷脂对实验结果有一定的影响,常用的C18和PSA对磷脂的去除效率不高,加大C18和PSA的量会降低一部分兽药的回收率。利用磷酸基团与锆的强配位作用,在净化剂中加入二氧化锆微球,可高效除去提取液中的磷脂,这样既可以避免因加入过量C18和PSA降低回收率,同时又可消除磷脂带来的基质影响。结果如图3所示,加入二氧化锆后,三种化合物的峰面积有了明显的提高,说明二氧化锆的加入有效吸附了基质中的磷脂,减小了基质抑制效应带来的影响。随着二氧化锆加入量的增大,三种化合物的峰面积基本维持不变,因为所称取的鱼肉质量有限,提取出的磷脂量有限,10 mg二氧化锆足以吸附提取液中的磷脂,同时考虑到各实验样品磷脂含量的差异性问题及实验成本,最终确定二氧化锆吸附剂的用量为20 mg。

图3 二氧化锆加入量对峰面积的影响Fig.3 Effects of different ZrO2 addition on the peak area of three nitromidazoles
2.4 基质效应
由图4可见,MNZ、HMMNI、DMZ的基质效应分别为-34.8%、-28.4%、-34.7%,三种化合物的基质效应均为负值,且绝对值均大于20%,说明基质对三种化合物存在明显的抑制作用,原因可能为,前处理过程中,鱼肉中的可溶性蛋白、多肽等内源性物质随目标物共同流出,在质谱分析电离过程中这些共同流出物抑制了目标物的离子化。由于无法获得与检测化合物一一对应的同位素内标物,因此本文采用更为常见的基质配标准曲线定量的方法,来降低基质效应的影响。

图4 三中硝基咪唑的基质效应Fig.4 Matrix effects of the three nitromidazoles
2.5 线性方程、检出限和定量限
如表2所示,三种化合物的在0.5~20 ng/mL范围内线性关系良好,R2均大于0.998;MNZ和DMZ的检出限为0.2 μg/kg,HMMNI的检出限为0.5 μg/kg,MNZ和DMZ的定量限为 0.6 μg/kg,HMMNI的定量限为1.2 μg/kg。

表2 三种硝基咪唑的线性方程、线性范围、检出限、定量限Table 2 Linear equations linear relationships,LODs,LOQs of three nitromidazoles
2.6 回收率和精密度
如表3所示,三种硝基咪唑在三个加标水平下的回收率在88.1%~107.0%之间,相对标准偏差在2.43~8.17%之间。方法具有较高的准确度和精密度,符合实际检测要求。
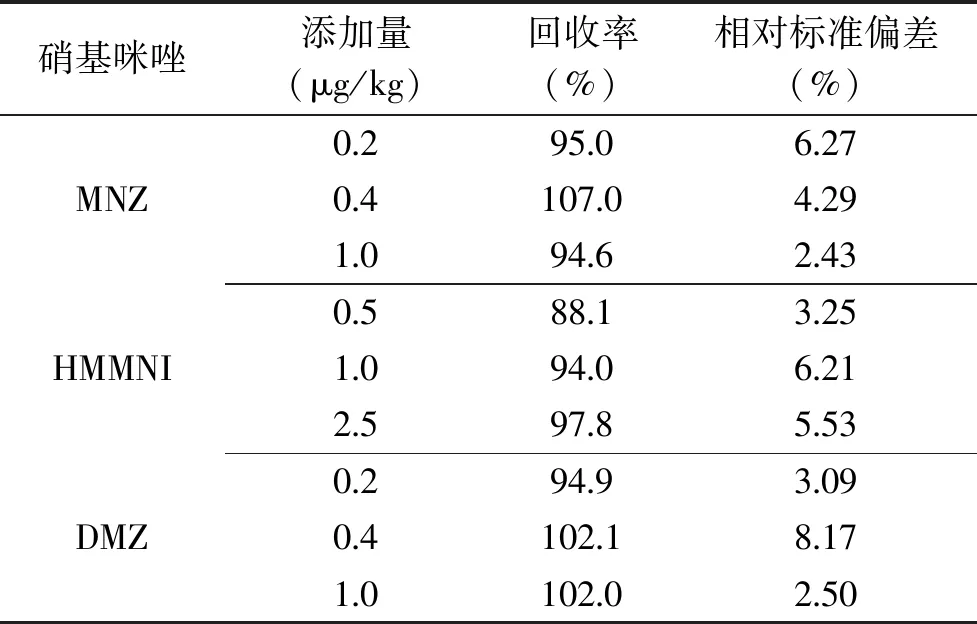
表3 三种硝基咪唑的加标回收率及相对标准偏差(n=6)Table 3 Recoveries and relative standard deviations of three nitromidazoles(n=6)
2.7 市售样品检测
按照优化后的处理方法及色谱条件,对本地不同超市市售的50批次鱼样品进行DMZ、HMMNI及MNZ的检测。实验结果显示,只有1批次的鳗鱼检出MNZ,其含量为0.7 μg/kg,其余产品均未检出这三种硝基咪唑类药物。
3 结论
本文针对动物源性产品中磷脂含量高的基质特点,结合已有的研究及行业标准,通过加入二氧化锆,改进了传统的QuEChERs方法以消除鱼肉样品中磷脂带来的基质干扰,并优化仪器分析参数、样品前处理及二氧化锆加入量。与传统的QuEChERs相比,分析方法的选择性及灵密度有所提高,其检出限在0.2~0.5 μg/kg之间,定量限为0.6~1.2 μg/kg之间;平均回收率在88.1%~107.0% 范围内,相对标准偏差在 2.43%~8.17%范围内。该方法具有准确、高效、灵敏度高的优点,适用于鱼肉中DMZ、HMMNI及MNZ的快速检测和日常筛查。虽然本文只选取了两种硝基咪唑及一种其代谢产物作为目标分析物,但文中的前处理方法可能适用于具有相似分子结构的其他类硝基咪唑类药物,乃至其他兽残类药物的检测。
