·生物技术与方法·地衣芽胞杆菌FLP/FRT基因编辑系统的构建及验证
2019-03-27李宗文李由然顾正华丁重阳张梁徐沙石贵阳
李宗文,李由然,顾正华,丁重阳,张梁,徐沙,石贵阳
1 江南大学 粮食发酵工艺与技术国家工程实验室,江苏 无锡 214122
2 江南大学 生物工程学院,江苏 无锡 214122
地衣芽胞杆菌Bacillus licheniformis是一种常见的革兰氏阳性细菌,具有耐热、酶系丰富、产酶量高和安全等诸多优良特性,是最具应用潜力和价值的芽胞杆菌之一。目前地衣芽胞杆菌被广泛用于各种蛋白质的胞外表达,包括α-淀粉酶、蛋白酶、脂肪酶、青霉素酶等,产量可达25 g/L[1]。此外,地衣芽胞杆菌在饲料添加、医药、环境治理以及农业病害防治等领域也有重要应用[2]。
当前以基因组序列信息为蓝图,基于人工设计的遗传扰动而进行的代谢工程已成为研究地衣芽胞杆菌复杂代谢途径以及进行菌种改造构建相关表型的有效策略。然而,受限于地衣芽胞杆菌遗传转化效率不高[3],对其进行基因操作存在一定困难,目前文献报道的几种基因编辑方法在使用效果上存在多方面的不足。传统的转化敲除盒片段同源替换目标基因的方法是实现微生物基因敲除的经典策略,主要通过微生物本身的RecA重组系统 (主要包括RecA和RecBCD等蛋白) 发挥作用[4]。但是受限于地衣芽胞杆菌遗传转化效率不高,且外源DNA受到限制修饰系统的降解[5],导致该方法运用到地衣芽胞杆菌中敲除效率非常低,而且得到的阳性转化子残留抗性标记[6]。CRISPR/Cas9系统是近年来基因编辑的研究热点,有报道运用CRISPR/Cas9n (Cas9的突变体,单链切割) 系统对地衣芽胞杆菌yvmC基因进行敲除且效率最高可达100%[7]。该方法需要将Cas9n基因表达盒整合到基因组上。还有文献报道利用温敏质粒介导同源双交换来失活目标基因[8-10]。Nahrstedt等[8]以温敏质粒pE194为载体构建敲除质粒敲除了地衣芽胞杆菌DNA修复相关基因recA和芽胞形成关键基因spoIV来研究解决生物污染问题。该方法的优点是把基因敲除分成转化和重组两个独立事件,转化成功后首先大量扩增转化子,之后再进行敲除重组过程,因此可以克服传统方法因遗传转化效率低导致敲除困难的问题,不足之处是由于敲除盒上没有设计抗性标记,会造成筛选工作量很大。因此,综合上述关于地衣芽胞杆菌基因编辑方法的现状,主要有三点不足:一是受限于转化效率不高导致的敲除困难;二是筛选量大;三是残留抗性标记。
FLP/FRT重组系统是发现于酿酒酵母2 µm环状质粒上的位点特异性重组 (Site-specific recombination) 系统,通过FLP重组酶 (Flippase recombination enzyme) 特异性识别FRT位点(FLP recombination target) 可介导2个FRT位点之间DNA片段的删除、倒位与置换等[11-12]。FLP/FRT重组系统由于具有较高的重组效率和靶向性,已经被广泛应用于细菌、酵母等微生物以及拟南芥、水稻、小鼠、果蝇、线虫等高等真核模式生物的研究中,实现了基因敲除、敲入、点突变、缺失突变、染色体组大片段删除等基因工程操作[13-15]。Sanchez-Martinez等[14]应用FLP/FRT系统实现在白假丝酵母Candida albicans中删除抗性标记基因URA3和整合外源基因到基因组上。
本研究借助温敏质粒介导基因敲除的策略,在地衣芽胞杆菌中构建FLP/FRT基因编辑系统,利用温敏质粒介导敲除的方法克服敲除效率低的问题,敲除盒中间设计抗性标记解决筛选量大的问题,利用FLP/FRT系统的特异性重组作用解决抗性标记残留问题,并以此成功敲除α-淀粉酶基因amyL、蛋白酶基因aprE以及敲入外源透明颤菌血红蛋白基因vgb,为地衣芽胞杆菌的遗传改造提供良好的方法参考。
1 材料与方法
1.1 材料
1.1.1 菌株及质粒
实验所用菌株及质粒见表1。
1.1.2 主要工具酶、试剂和培养基
文中所用的2×TaqPCR Master Mix、2×PfuPCR Master Mix购自杭州宝赛公司;Fast DigestedTM快速限制性内切酶购自美国Thermo公司;T4 DNA连接酶和pMD19T-simple购于大连TaKaRa公司;卡那霉素、氨苄青霉素、四环素购自Sigma公司;质粒DNA小剂量提取试剂盒、DNA纯化试剂盒和DNA凝胶回收试剂盒购自北京博大泰克生物有限公司;蛋白胨、酵母粉、琼脂粉购自OXOID公司;其他试剂均为国产或进口分析纯。大肠杆菌和地衣芽胞杆菌生长用LB培养基(g/L):蛋白胨10,酵母粉5,NaCl 10。配置固体LB时加入1.5%–2.0%的琼脂粉。在菌株构建和摇瓶发酵过程中,培养基中添加的氨苄青霉素、卡那霉素、四环素终浓度分别为100、30、20 μg/mL。
1.1.3 引物
实验所用引物见表2,引物均由苏州金唯智生物科技有限公司合成。
1.2 方法
1.2.1 敲除及敲入质粒的构建
敲除质粒的构建过程如下所述。本研究选取地衣芽胞杆菌9945a α-淀粉酶 (α-amylase) 基因amyL(CP005965 REGION: 723302..724840) 和蛋白酶 (Subtilisin Carlsberg) 基因aprE(CP005965 REGION: 1207540..1208679) 为敲除试验基因。以9945a基因组为模板,分别利用引物amyLXhoⅠ-F/amyL-Hind Ⅲ-R 及aprE-XhoⅠ-F/aprESmaⅠ-R克隆出α-淀粉酶基因amyL及蛋白酶基因aprE,连接到pMD19T-simple上,得到质粒19T-amyL及19T-aprE。以质粒pMA5为模板,利用引物FRT-KpnⅠ-Kan-F/FRT-SalⅠ-Kan-R进行PCR扩增,得到首尾两端含同向FRT位点的卡那霉素 (Kanamycin,Kan) 抗性基因表达元件并用KpnⅠ和SalⅠ双酶切,将酶切产物与经相同双酶切的19T-amyL大片段连接,构建得到19T-AFKF(敲除盒片段amyL-FRT-Kan-FRT-amyL,简称AFKF),左右同源臂长分别为420 bp和547 bp。以19T-aprE为模板,利用引物aprE-KpnⅠ-F/aprE-SalⅠ-R反向PCR (引物结合在aprE基因的中部位置,反向PCR可得到左右同源臂),PCR产物用KpnⅠ和SalⅠ酶切纯化后与卡那霉素抗性基因表达元件连接,构建得到19T-EFKF (敲除盒片段aprE-FRT-Kan-FRT-aprE,简称EFKF),左右同源臂长分别为513 bp和409 bp。最后用XhoⅠ和Hind Ⅲ酶切19T-AFKF,胶回收敲除盒片段AFKF,连接到相同酶切的pNZTT质粒,得到amyL基因的温敏敲除质粒pNZTT-AFKF;XhoⅠ和SmaⅠ酶切19T-EFKF,胶回收敲除盒片段EFKF,连接到相同酶切的pNZTT质粒,得到aprE基因的温敏敲除质粒pNZTT-EFKF。
敲入质粒的构建过程如下所述。文中选取了丙酮酸甲酸裂解酶 (Putative formate C-acetyltransferase)基因pflB(CP005965 REGION: 2135016..2137240)为敲入位点,整合外源透明颤菌血红蛋白 (Vitreoscillahemoglobin) 基因vgb。首先以9945a基因组为模板,利用引物pflB-SmaⅠ-F/pflB-XhoⅠ-R扩增得到pflB基因,连接到pMD19T-simple,构建质粒19T-pflB。以19T-pflB为模板,利用引物pflB-PstⅠ-R/pflB-BamHⅠ-F反向PCR (引物结合在pflB基因的中部位置,反向PCR可得左右同源臂),扩增产物经PstⅠ和BamHⅠ酶切后待用;以pHY300-PLK质粒为模板,利用引物FRT-BamHⅠ-Tet-F/FRTPstⅠ-Tet-R扩增得到两端含同向FRT位点的四环素 (Tetracycline,Tet) 抗性基因表达元件,经BamHⅠ和PstⅠ酶切后与上面的反向PCR产物连接,构建质粒19T-PFTF。以pWB980-vgb为模板,利用引物vgb-BamHⅠ-F/vgb-BamHⅠ-R扩增得到含P43启动子的vgb表达盒,BamHⅠ单酶切后连接到相同单酶切的19T-PFTF,构建得到质粒19T-PFTF-vgb(敲入盒片段pflB-vgb-FRT-Tet-FRT-pflB,简称PFTF-vgb),左右同源臂长分别为935 bp和1 003 bp。最后用SmaⅠ和XhoⅠ酶切19T-PFTF-vgb得到敲入盒PFTF-vgb,连接到相同酶切的pNZTK质粒,得到温敏敲入质粒pNZTK-PFTF-vgb。

表2 文中所用引物Table 2 Primers used in this study
1.2.2 FLP重组酶表达质粒的构建
以质粒pCP20为模板,利用引物flp-SmaⅠ-F/flp-SmaⅠ-R扩增出FLP重组酶结构基因flp,扩增产物flp经SmaⅠ单酶切后纯化待用。质粒pMA5经NdeⅠ酶切纯化后用等量的2×PfuDNA聚合酶72 ℃孵育15 min补平粘性末端,然后与flp连接,构建中间质粒pMA5-flp。以pMA5-flp为模板,利用引物flp-pMA-SmaⅠ-F/flp-pMAXhoⅠ-R扩增得到含HpaⅡ启动子的flp表达盒,flp表达盒与pNZTT经过相同的SmaⅠ和XhoⅠ酶切后连接得到pNZTT-flp,与pNZTK经相同的SmaⅠ和XhoⅠ酶切后连接得到pNZTK-flp。为行文方便,称pNZTT-flp为敲除消抗质粒,称pNZTK-flp为敲入消抗质粒。
1.2.3 基因敲除与基因敲入
基因敲除步骤如下所示。以敲除amyL为例,把构建好的敲除质粒pNZTT-AFKF通过电转的方法转入地衣芽胞杆菌9945a细胞中。电转方法参考李由然[16],特别地,种子需经LB平板活化,菌体要充分洗涤4次以尽可能除去离子以及质粒加入量不少于200 ng。转化成功的9945a/pNZTTAFKF首先在30 ℃、200 r/min下增殖活化,形成足够的新鲜菌浓;转接300 μL菌液至新的15 mL LB培养基,42 ℃、250 r/min无抗培养1代,培养时间约14–20 h;用接种环蘸取菌液在卡那霉素抗性平板上划线,平板置于37 ℃培养箱至长出单菌落,经PCR验证得到单交换菌株;单交换菌株先在37 ℃摇瓶扩增,后转接150 μL单交换菌液至新的15 mL LB培养基,30 ℃、200 r/min无抗培养2代,每代培养时间约24–30 h;最后用无菌水稀释10–5–10–6涂布卡那霉素抗性平板,平板置于37 ℃培养箱培养至长出单菌落。挑取一定数量的单菌落在卡那霉素抗性平板上划线扩增,然后一一对应划线至四环素抗性平板,在四环素抗性平板上不长的单菌落符合敲除重组子抗性特征,用敲除验证引物amyL-F/amyL-M-R、amyL-M-F/amyL-R进行菌落PCR验证。同样的流程用温敏敲除质粒pNZTT-EFKF对蛋白酶基因aprE进行敲除。
基因敲入的原理是在敲除质粒的基础上在同源臂和同侧FRT位点之间插入外源基因,然后经过与敲除相同的操作流程,可实现外源基因整合至基因组。具体为:把构建好的敲入质粒pNZTKPFTF-vgb通过电转的方法转入地衣芽胞杆菌9945a中,然后经过与敲除相同的变温传代过程,最后抗性平板筛选出具有卡那霉素抗性,同时对四环素敏感的,并用敲入验证引物pflB-F/pflB-M-R、pflB-M-F/pflB-R进行菌落PCR验证。
1.2.4 抗性回收
前面得到的敲除重组子与敲入重组子基因组上都插入了抗性标记基因,使得菌体带有抗性。抗性回收策略是通过电转的方法向重组子中导入一个FLP重组酶表达质粒,表达的FLP重组酶特异性识别FRT位点并介导2个FRT位点之间抗性基因的删除。其中敲除重组子导入pNZTT-flp,敲入重组子导入pNZTK-flp,然后在30 ℃、200 r/min条件下培养2代,最后抗性平板负筛出抗性丢失的重组菌,并用相应引物进行菌落PCR鉴定并测序验证。
抗性回收后携带消抗质粒的重组菌在37 ℃下无抗摇瓶传代2次即可丢失质粒。
1.2.5 α-淀粉酶及蛋白酶酶活检测
淀粉酶酶活的测定参照Kim等[17]的方法进行。酶活单位 (U) 定义为:在40 ℃、pH 6.5的反应条件下,1 mL酶液每小时分解可溶性淀粉生成麦芽糖的μmol数。
蛋白酶酶活采用Folin 酚法[18]测定。酶活单位(U) 定义为:在40 ℃、pH 10.0条件下,1 mL酶液每分钟水解酪素产生1 μg酪氨酸为一个酶活单位。
1.2.6 vgb整合表达检测
运用细菌总RNA提取试剂盒提取9945a.4总RNA,然后逆转录成cDNA,再设计vgb特异性引物进行荧光定量PCR (real-time PCR, RT-PCR)分析检测,以核糖体S5蛋白基因rpsE[19-20]为内参,根据基因达到荧光阈值的循环数 (Ct值),采用2–△Ct法计算vgb基于内参基因rpsE的相对表达量。
2 结果与分析
2.1 敲除质粒及敲入质粒的构建
将构建得到的敲除质粒pNZTT-AFKF用Hind Ⅲ和XhoⅠ酶切,结果如图1A所示,所得条带大小与理论值2 209 bp和5 672 bp相符,表明质粒构建正确;pNZTT-EFKF用SmaⅠ和XhoⅠ酶切并进行核酸胶电泳,结果如图1B所示,两条带大小与理论值2 175 bp和5 329 bp相符,表明质粒构建正确。
类似地,将构建得到的敲入质粒pNZTKPFTF-vgb用Hind Ⅲ和XhoⅠ酶切,结果如图1C所示,目的条带大小与理论值3 320 bp和6 183 bp相符,表明质粒构建正确。
所有构建得到的重组质粒经测序验证无序列突变。
2.2 FLP重组酶表达质粒的构建
构建得到的FLP重组酶表达质粒pNZTT-flp经SmaⅠ和XhoⅠ酶切,结果如图2A所示,条带大小与理论值1 760 bp和5 329 bp相符,表明敲除消抗质粒pNZTT-flp构建正确。flp表达盒经测序验证无序列突变。
同理,pNZTK-flp用限制性内切酶SmaⅠ和XhoⅠ酶切,结果如图2B所示,两条带大小与理论值1 760 bp和4 852 bp相符,表明敲入消抗质粒pNZTK-flp构建正确。flp表达盒经测序验证无序列突变。
2.3 amyL及aprE基因的敲除及抗性回收
文中所用的pNZT1质粒是温度敏感型滚环复制质粒,属于大肠杆菌-芽胞杆菌穿梭质粒,培养温度在30 ℃及以下时可稳定复制,37 ℃及以上质粒丢失[21]。pNZT1携带有红霉素抗性基因,但由于红霉素对文中宿主菌实际应用效果不好,因此在多克隆位点的NotⅠ处插入卡那霉素抗性基因表达盒得到pNZTK质粒,插入四环素抗性基因表达盒得到pNZTT质粒。插入的抗性基因作为载体自身的抗性使用。
基因敲除原理是两步同源重组作用[9,21-22]。以敲除amyL为例,9945a/pNZTT-AFKF首先在30 ℃下增殖活化,目的是为了获得生长旺盛的含敲除质粒的菌液及足够的菌浓;菌液转接到42 ℃无抗培养,此温度下质粒无法复制,敲除质粒携带的一侧同源臂和amyL基因的同源序列之间发生同源重组,使得整个敲除质粒线性整合到基因组上,此过程称为单交换,单交换菌株具有卡那霉素和四环素双重抗性。实验结果显示此步骤也有可能直接得到双交换菌株。得到的单交换菌株转接至30 ℃下无抗培养,此时低温诱导复制子发生滚环复制使质粒部分从基因组上切下,同时敲除盒的另一侧同源臂和amyL基因的同源序列之间发生同源重组,此过程称为双交换,双交换菌株只有卡那霉素抗性。通过两次同源交换,amyL基因被敲除质粒pNZTT-AFKF上的敲除盒同源替换,导致amyL缺失及插入失活。敲除原理[21]及抗性回收示意图见图3。
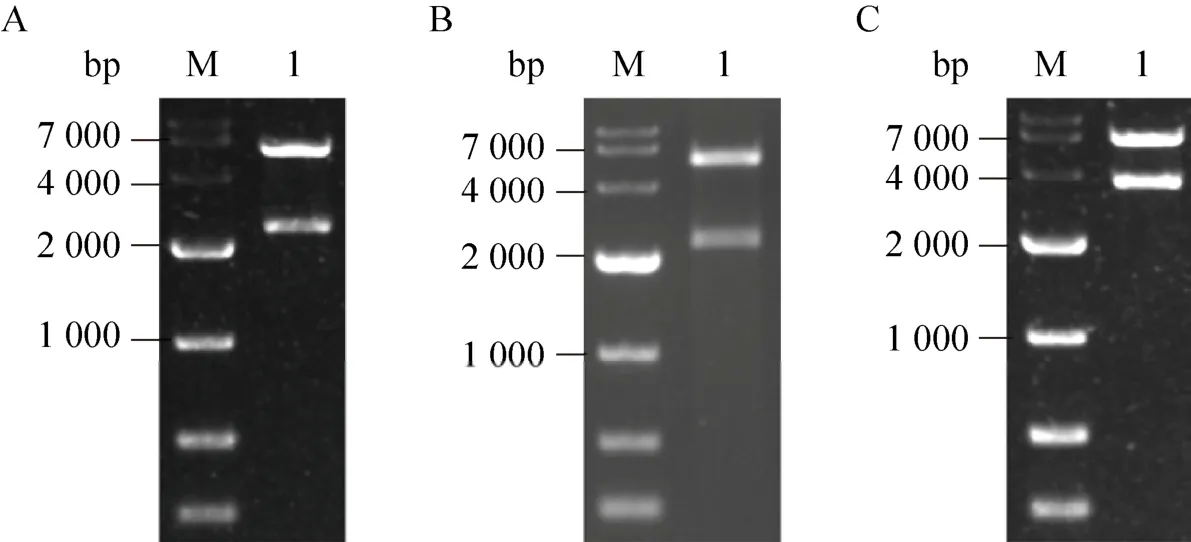
图1 敲除质粒pNZTT-AFKF (A)、pNZTT-EFKF (B) 及敲入质粒pNZTK-PFTF-vgb (C) 的酶切验证Fig.1 Identification of knock-out vector pNZTT-AFKF (A), pNZTT-EFKF (B) and knock-in vector pNZTK-PFTF-vgb(C).(A) M: DL 10 000 DNA marker; lane 1: digested by XhoⅠ and Hind Ⅲ.(B) M: DL 10 000 DNA marker; lane 1:digested by SmaⅠand XhoⅠ.(C) M: DL 10 000 DNA marker; lane 1: digested by XhoⅠand Hind Ⅲ.

图2 敲除消抗质粒pNZTT-flp (A) 及敲入消抗质粒pNZTK-flp (B) 的酶切验证Fig.2 Identification of marker recycling vectors pNZTT-flp (A) and pNZTK-flp (B).M: DL 10 000 DNA marker; lane 1: digested by XhoⅠand SmaⅠ.
实验结果显示,敲除质粒pNZTT-AFKF通过单交换方式线性整合至9945a基因组上属于高概率事件,效率高达90%以上。取低温培养后的双交换菌液用无菌水稀释10–6涂卡那霉素抗性平板,从长出的单菌落中均匀挑取28个单菌落,四环素抗性平板负筛到22株对四环素敏感,表明双交换的效率为78.6% (图4)。从中任意挑8株用敲除验证引物amyL-F/amyL-M-R和amyL-M-F/amyL-R进行菌落PCR验证,结果如图5A所示,PCR条带大小均和理论相符,而原始菌没有条带,说明amyL基因被成功敲除,敲除重组子命名为9945a.1。从中任意挑选一株9945a.1提基因组作为模板,用引物amyL-F/amyL-R进行PCR扩增,扩增产物测序,测序结果和理论相符。

图3 amyL敲除原理[21]及FLP/FRT系统介导的抗性回收示意图Fig.3 Scheme of deletion amyL[21] and marker recycling by FLP/FRT system.Note that the initial recombination can occur either in the upstream portion of the target gene as shown here, or at the downstream portion.In both cases the final structure of the chromosome is the same.

图4 amyL敲除双交换抗性筛选Fig.4 The resistance screening of double crossover for amyL knock-out.(A) Kanr plate.(B) Tetr plate.The colonies between plate A and plate B are corresponding one by one.
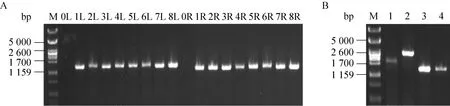
图5 amyL突变株9945a.1和9945a.2的菌落PCR验证Fig.5 Identification of the 9945a.1 and 9945a.2 by colony PCR.(A) M: molecular mass marker; 0L–8L: PCR products using primers amyL-F/amyL-M-R; 0R–8R: PCR products using primers amyL-M-F/amyL-R.Among the lanes, 1L–8L and 1R–8R took genome of 9945a.1 as a template, 0L and 0R act the contrast whose templates were 9945a.The sizes of PCR products are shown in Fig.3.(B) M: molecular mass marker; lane 1: PCR product of 9945a using primers amyL-F/amyL-R, 1 901 bp;lane 2: PCR product of 9945a.1 using primers amyL-F/amyL-R, 2 603 bp; lane 3 and lane 4:PCR products of 9945a.2 using primers amyL-F/amyL-R, 1 417 bp.
对9945a.1进行抗性回收,在四环素抗性平板上挑选了30个单菌落,一一对应划线至卡那霉素平板上筛选到24株卡那霉素抗性丢失,抗性回收效率高达80%。任意挑选2株对卡那霉素敏感的菌落用引物amyL-F/amyL-R进行PCR验证,结果见图5B,抗性回收后条带大小为1 417 bp,明显小于原始菌的1 901 bp和抗性回收前的2 603 bp,说明抗性回收成功。抗性回收后的敲除重组菌命名为9945a.2。任意挑取1株9945a.2提基因组作为模板,用引物amyL-F/amyL-R进行PCR扩增,扩增产物测序,测序结果和理论相符。
9945a.2含有质粒pNZTT-flp,经过37 ℃无抗传代2次可得到质粒丢失的无抗单菌落。
为了进一步验证敲除系统的可行性及敲除效率,还对9945a.2进行了蛋白酶基因aprE的敲除。aprE的敲除原理和操作流程与amyL相同,第一步高温培养下pNZTT-EFKF通过单交换方式线性整合至基因组,此步骤效率高于50%;第二步低温培养诱导单交换菌株发生双交换,从挑选的18株单菌落中筛选到14株具有卡那霉素抗性而对四环素敏感 (图6A和6B),表明双交换效率为77.7%。对双交换成功的aprE敲除重组子命名为9945a.3。同样地,对9945a.3通过电转方法导入消抗质粒pNZTT-flp进行了抗性回收,并用引物aprE-XhoⅠ-F/aprE-SmaⅠ-R进行菌落PCR鉴定,结果如图6C所示,原始菌条带大小为1 116 bp,消抗前为2 185 bp,抗性回收后为 999 bp。抗性回收后的重组菌命名为9945a.4。

图6 aprE敲除双交换抗性筛选及抗性回收PCR验证Fig.6 The resistance screening of double crossover for aprE knock-out and identification of marker recycling by PCR.(A) Kanr plate.(B) Tetr plate.The colonies between plate A and plate B are corresponding one by one.(C) M: DL 10 000 DNA marker; lane 1: PCR product of 9945a, 1 116 bp; lane 2: PCR product of 9945a.3, 2 185 bp; lane 3: PCR product of 9945a.4, 999 bp.All PCR products used primers aprE-XhoⅠ-F/aprE-SmaⅠ-R.
2.4 vgb基因的敲入及抗性回收
基因敲入的原理是在敲除质粒的基础上在同源臂和同侧FRT位点之间插入外源基因,然后经过与敲除相同的重组过程,最后可实现外源基因在基因组上的定点敲入。结果显示,敲入质粒pNZTK-PFTF-vgb通过单交换线性整合至9945a基因组的概率高达95%以上,属于非限速步骤。取低温培养后的双交换菌液稀释10–6涂四环素平板,从长出的单菌落中均匀挑取了30个单菌落,经筛选有27株对卡那霉素敏感,如图7所示,双交换的效率为90%。从中任意挑8株用敲入验证引物pflB-F/pflB-M-R和pflB-M-F/pflB-R进行菌落PCR验证,结果如图8A所示,8株菌均成功敲入,即pflB基因被敲除,同时vgb整合至pflB位点。vgb基因敲入重组子命名为9945a.5。

图7 vgb敲入双交换抗性筛选Fig.7 The resistance screening of double crossover for vgb knock-in.(A) Tetr plate.(B) Kanr plate.
任意挑选1株9945a.5导入消抗质粒pNZTKflp进行抗性回收,筛选得到对四环素敏感的单菌落,用引物pflB-F/pflB-R进行菌落PCR,结果如图8B所示。抗性回收前扩增产物长5 101 bp,回收后为3 525 bp,图8B条带小于4 000 bp,表明抗性回收成功,抗性回收后的重组菌命名为9945a.6。
2.5 α-淀粉酶及蛋白酶酶活检测
amyL突变株9945a.2 α-淀粉酶酶活检测以地衣芽胞杆菌9945a原始菌为对照,结果如图9A所示,9945.2的淀粉酶比酶活只有原始菌的4.7%左右,酶活大幅度减少,这也证实了amyL基因被敲除。据文献分析,残余的酶活可能是来自其他淀粉酶比如Taka-淀粉酶 (Taka-amylase)[23]或普鲁兰酶[16]。aprE敲除后蛋白酶比酶活由原始菌的16.3 U/mg减少到3.2 U/mg,减少了80.4%;淀粉酶基因的敲除对菌株产蛋白酶几乎无影响。
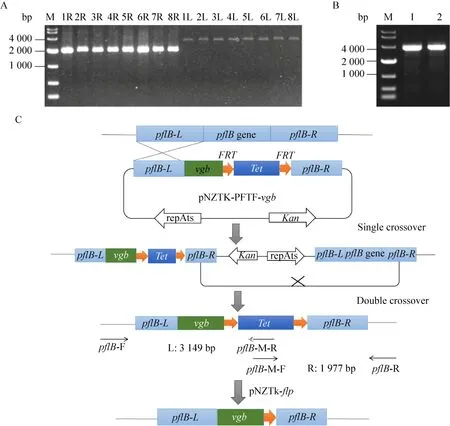
图8 vgb敲入突变株PCR验证及敲入策略示意图Fig.8 Identification of the 9945a.5 and 9945a.6 by PCR and the strategy of knocking in vgb.(A) M: DL 10 000 DNA marker; 1L–8L: PCR products of 9945a.5 using primers pflB-F/pflB-M-R; 1R–8R: PCR products of 9945a.5 using primers pflB-M-F/pflB-R.The sizes of PCR products are shown in Fig.8C.(B) M: DL 10 000 DNA marker; lane 1 and lane 2: PCR products of 9945a.6 using primers pflB-F/pflB-R, 3 525 bp.(C) Brief scheme of knocking in vgb and marker recycling by FLP/FRT system.Note that basic principle of knocking in a heterogenous gene is the same with deletion a gene like amyL, except for carrying a heterogenous gene between a homology arm and the same side FRT.
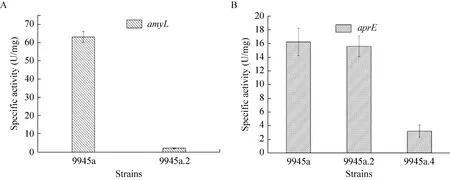
图9 敲除突变株α-淀粉酶 (A) 及蛋白酶 (B) 酶活检测Fig.9 Activities of α-amylase (A) and protease (B) of the knock-out mutants and wide type.
2.6 vgb表达检测
vgb基因编码透明颤菌血红蛋白VHB,VHB受氧浓度调控,在贫氧条件下可维持菌的生长。vgb的RT-PCR检测结果如表3所示,9945a.6中检测到vgb的表达,且相对rpsE的表达量为1.23,表达水平相对较高,这可能是由于vgb的启动子P43是组成型的强启动子,而原始菌9945a中未检测到vgb的表达,说明外源的vgb成功整合至基因组且得到表达。

表3 vgb敲入菌株9945a.6及原始菌rpsE和vgb表达水平分析Table 3 Relative mRNA abundance of rpsE and vgb in 9945a.6 and the wide type
3 讨论
地衣芽胞杆菌作为极具应用潜力的工业菌种之一,限制其应用进一步拓展的因素之一就是基因操作困难。转化敲除盒片段直接进行双交换的一步法在转化效率不高的条件下很难得到阳性转化子。本研究借鉴了温度敏感型质粒进行基因敲除的成功案例,首次结合运用FLP/FRT重组系统,构建出一套适用于地衣芽胞杆菌的、更加实用的基因编辑系统。运用该系统高效敲除了地衣芽胞杆菌α-淀粉酶基因amyL及蛋白酶基因aprE;实现了外源基因vgb在基因组上的成功敲入与表达。本方法是对已有温敏质粒介导基因敲除技术的有机延伸与拓展。原技术由于插入位点不带抗性标记,需要多步传代富集阳性转化子,且依靠无抗平板初筛,工作量大。本方法在预先引入FRT重组位点的前提下,利用设计于插入位点的抗性标记,显著缩短了阳性转化子筛选周期及减少筛选工作量;结合FLP重组酶介导的高效率抗性回收,同样达到了无抗性标记残留的基因编辑。在地衣芽胞杆菌常用抗生素有限的背景下,仅需单一抗性标记即可实现多基因的连续敲除。
另一方面,该系统还存在一个Cre-loxP和FLP/FRT系统普遍存在的问题,即每一次运用该系统进行基因敲除,都会在基因组上留下1个FRT识别位点的疤痕,因此如果对基因组进行多轮操作,留下的多个识别位点之间可能会发生重组,降低基因组的稳定性。为了避免这种情况,后续可参考Yan等[24]使用突变型的FRT位点[25],从而提高遗传稳定性。
总体来说,文中构建的FLP/FRT基因编辑系统能够有效地在地衣芽胞杆菌中敲除目标基因以及敲入外源基因,为地衣芽胞杆菌的代谢改造及基因功能研究提供了良好的实验方法。有文献表明[21-22],温敏质粒介导的基因敲除方法也可运用到遗传转化困难的其他芽胞杆菌中,因此可推测本研究成果在芽胞杆菌中具有一定的通用性。
