利用CRISPR-Cas9系统构建新型异戊酰螺旋霉素Ⅰ产生菌
2019-03-27张晓婷张妍戴剑漉王以光赫卫清
张晓婷,张妍,戴剑漉,王以光,赫卫清
1 中国医学科学院 医药生物技术研究所 卫健委抗生素生物工程重点实验室,北京 100050
2 沈阳同联集团有限公司,辽宁 沈阳 110042
必特螺旋霉素 (Bitespiramycin,BT) 是将耐热链霉菌Streptomyces thermotolerans的4″-异戊酰基转移酶基因 (4″-Isovaleryltransferase gene,ist) 导入到螺旋霉素 (Spiramycin,SP) 产生菌螺旋链霉菌Streptomyces spiramyceticus中进行表达,所产生的发酵产物[1]。BT的主组分是异戊酰螺旋霉素(Isovalerylspiramycin,ISP) Ⅰ、Ⅱ和Ⅲ,其结构式如图1所示。因为SP的3位酰基化酶的底物特异性不强,可以同时识别乙酰基和丙酰基底物,导致出现3种主组分。3位只是羟基的为Ⅰ组分,Ⅱ组分是3位羟基乙酰化产物,Ⅲ组分是丙酰化产物。BT是多组分药物,在发酵产物提纯和质控方面难度较大。经研究初步证实ISP Ⅰ的药物活性与BT相当,因此可以作为一种单组分药物进行后续开发。前期研究中通过阻断SP的3位酰基化酶基因bsm4,获得只产SPⅠ的基因工程菌株[2],再导入2个拷贝的ist基因[3],得到只产ISP Ⅰ的工程菌株[4]。但此菌株ISP Ⅰ的产量很低,发酵液的效价只有200 μg/mL。为了提高ISP Ⅰ的发酵产量,又将ist基因和其正调控基因acyB2整合到ISP Ⅰ菌株的染色体上,通过育种筛选,获得效价在800 μg/mL左右的高产菌株S.spiramyceticusWSJ-IA[5–6]。此高产菌株中已经包含硫链丝菌素和阿普拉霉素两种抗性基因,利用现有的链霉菌质粒很难再对其进行定向遗传操作。因此采用等离子诱变和传统诱变相结合的方法对WSJ-IA菌株进行诱变育种,获得效价在2 000 μg/mL左右的高产菌株[7],基本满足大型发酵的要求。此菌株共进行3次遗传操作,特别是将bsm4基因进行部分删除后,菌株的产量下降为原株的50%左右。在菌株染色体的2个不同位置中还包含3个ist基因,在传代过程中有发生同源重组的可能性,不利于菌株的遗传稳定。
为了获得遗传更加稳定的ISP Ⅰ菌株,利用近几年新出现的CRISPR-Cas9基因编辑系统[8-9],可以方便地对靶基因进行阻断或替换。本研究拟将组成型强启动子ermEp*控制下的ist基因替换bsm4基因,从而获得新的ISP Ⅰ菌株。
1 材料与方法
1.1 材料
1.1.1 菌种与质粒
螺旋霉素产生菌S.spiramyceticus1941由本实验室保存。大肠杆菌Escherichia coliDH5α感受态细胞、基因克隆和重组质粒构建的受体菌,购自北京全式金生物技术有限公司。pKC1139[10]链霉菌和大肠杆菌穿梭质粒,带有Apramycin抗性基因,本实验室保存。CRISPR-Cas9基因组编辑质粒pKCcas9do[11],由中国科学院上海生命科学研究院姜卫红研究员惠赠。pKCcas9do是以链霉菌温敏质粒pKC1139为出发质粒,在培养温度为37 ℃时停止复制。pKCcas9do质粒含有优化的cas9基因,由硫链丝菌素诱导的启动子tipA进行调控;人工合成的j23119启动子控制引导RNA(Single-guide RNA,sgRNA) 基因的转录;sgRNA由crRNA和tracrRNA组成,在crRNA中人工设计20 bp的靶结合序列,sgRNA 引导Cas9蛋白对靶位点进行切割。之后可以通过在37 ℃培养和传代,从宿主菌中去除此质粒,获得没有抗性而靶基因被改造的目的菌株。实验中用于构建质粒的引物序列见表1。

图1 异戊酰螺旋霉素的结构Fig.1 Structure of isovalerylspiramycin.
1.1.2 试剂
BT和ISP Ⅰ的对照品,本实验室自制。阿普拉霉素 (Apramycin,Am) 购自武汉远城科技发展有限公司。硫链丝菌素 (Thiostrepton,Tsr),购自Sigma公司。限制性内切酶、T4 DNA连接酶购自TaKaRa公司。KOD FX Neo DNA聚合酶 (TOYOBO) 购自北京天佑恒远生物科技有限公司。
1.1.3 培养基
S.spiramyceticus1941及其突变株的固体培养基 (g/100 mL):黄豆饼粉2,葡萄糖1,淀粉3,CaCO30.5,NaCl 0.4,琼脂粉1.5,自然pH值。1 μg/mL的Tsr诱导Cas9基因表达进行定点切割。R2YE培养基用于S.spiramyceticus1941的原生质体的制备和转化[12]。
S.spiramyceticus1941及其突变株的种子和发酵培养基 (g/100 mL) 详见文献[13]。
S.spiramyceticus1941及其突变株的生物检定培养基 (g/L):牛肉膏3.0,酵母膏3.0,蛋白胨(F 403)10.0,葡萄糖1.0,氯化钠5.0,琼脂12.0,pH 8.0。
1.2 方法
1.2.1 分子克隆
链霉菌总DNA的提取按文献[14]进行。E.coliDH5α与E.coliET12567感受态细胞的制备、分子克隆与鉴定按文献[15]进行。PCR反应采用KOD FX Neo DNA聚合酶反应体系。DNA测序由中美泰和生物技术 (北京) 有限公司完成。

表1 本文中所用引物Table 1 Primers used in this study
1.2.2 构建bsm4基因替换质粒
利用3对引物bsm4-LF/LR、ei-F/R和bsm4-RF/RR分别以S.spiramyceticus1941基因组DNA为模板进行扩增,获得左同源臂 (Left arm),ermEp*-ist和右同源臂 (Right arm) 3种PCR产物。将这些产物纯化后,进行重叠延伸PCR,获得 6.6 kb的目的片段,再进行XbaⅠ和HindⅢ酶切消化,与用同样酶切的pKC1139载体进行连接,得到pKC-ei重组质粒,如图2所示。
利用CRIPR-Cas9系统进行基因替换,先通过sgRNA-1和sgRNA-R引物扩增出引导Cas9进行特异性切割的引导序列sgRNA-1,用SpeⅠ和XbaⅠ进行双酶切后回收;利用sgRNA-2和sgRNA-R引物扩增出另一个引导序列sgRNA-2,也用SpeⅠ和XbaⅠ进行双酶切后回收;以pKC-ei为模板,利用Hist-F/R引物扩展出4.0 kb片段,用XbaⅠ和HindⅢ进行双酶切后回收,将sgRNA-1和sgRNA-2分别与4.0 kb片段克隆到pKCcasdo载体 (SpeⅠ/HindⅢ) 上,获得替换质粒pKcas-ei1和pKcas-ei2。
1.2.3 筛选目的菌株
将构建好的重组质粒通过原生质体转化法导入到S.spiramyceticus1941中,利用1 μg/mL Tsr诱导Cas9 表达,由sgRNA引导Cas9对靶位点进行切割,在质粒上有设计好的ermEp*-ist替代bsm4基因的同源片段。将发生同源双交换的目的菌株置于37 ℃培养,使质粒停止复制,之后通过Am和无抗性平板进行筛选,获得ermEp*-ist替代bsm4基因的目的菌株,进行PCR鉴定和测序验证。
1.2.4 发酵产物检测
含S.spiramyceticus1941转化子的斜面置于28 ℃培养 5–7 d,挖块至种子培养基 (50 mL培养基/250 mL三角瓶),28 ℃振荡 (200 r/min) 培养48 h,按1∶50的比例转接至发酵培养基中 (50 mL培养基/500 mL三角瓶),28 ℃振荡 (200 r/min)培养4 d,发酵液离心取上清液,用5.0 mol/L NaOH调至pH 8.5–9.0,加等体积乙酸乙酯萃取,8 mL萃取液离心浓缩至干燥,溶于200 μL乙腈,0.22 μm的滤膜过滤后,取3 μL进行HPLC检测 (日本岛津高效液相色谱仪,LC-10ATvp CLASS-VP V6.10):色谱柱为Kromasil C18,5 μm,4.6 mm×250 mm,流动相为乙腈/10 mmol/L pH 7.5–8.0的乙酸铵溶液 (65/35),流速为1 mL/min,检测波长231 nm。LC-MS (美国Thermo公司LTQ型液相色谱-质谱联用仪) 检测在本所仪器室进行,质谱条件:ES I源;正离子检测;鞘气 (N2) 流速0.3 L/min;辅助气 (He) 流速 0.1 L/min;源电压4.5 kV;毛细管温度350 ℃;毛细管电压4.5 V。
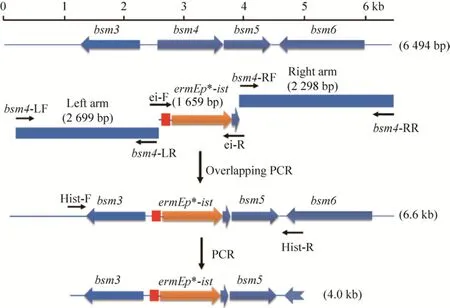
图2 bsm4基因替换质粒的构建Fig.2 Construction of bsm4 replacement plasmids.
2 结果与分析
2.1 bsm4上下游基因分析
BT产生菌S.spiramyceticusWSJ已经完成全基因组测序,并从序列中鉴定出的螺旋霉素生物合成基因簇 (GenBank登录号:MH 460451),3位的酰基转移酶基因命名为bsm4(图2),它的上游基因为bsm3,根据同源性推测其产物为23S rRNA甲基转移酶,参与核糖体23S rRNA的甲基化,与抗生素的抗性相关。下游基因bsm5为O-甲基转移酶基因,为螺旋霉素的后修饰基因,它与bsm4的开放阅读框 (Open reading frame,ORF)之间只有5 bp的基因间序列,推测二者共转录。要确保bsm5的正常转录,在使用ermEp*-ist替代bsm4部分序列时,不能在ist后面设计转录终止信号,同时需要保留bsm5翻译所需元件,如核糖体结合位点。因此在设计时保留了bsm4基因最后部分的111 bp序列。bsm6基因与bsm5的ORF方向相反,推测为crotonyl-CoA还原酶。ermEp*-ist替代bsm4后对其转录和翻译没有明显影响。
2.2 构建重组质粒
利用 3对引物bsm4-LF/LR、eiF/eiR和bsm4-RF/RR以S.spiramyceticus1941基因组DNA为模板进行PCR,分别得到2 699 bp的左同源臂、1 659 bp的ermEp*-ist片段和2 298 bp的右同源臂3种目的片段 (图3A),将这3种PCR产物通过重叠延伸PCR,获得3个片段拼接的产物 (6.6 kb)。泳道4和5是利用bsm4-LF/RR引物鉴定构建好的pKC-ei重组质粒,目的片段大小与预期相符 (图3B),并通过测序进行确证。将重组质粒pKC-ei通过原生质体转化的方法导入到S.spiramyceticus1941,利用自然发生的同源重组来获得ermEp*-ist替代bsm4的目的菌株。因为没有抗性标记只能利用传代和PCR进行筛选,经过几百株次的筛选仍然无法获得目的菌株。
之后利用CRISPR-Cas9系统的质粒pKCcas9do构建2种新的替换质粒pKcas-ei1和pKcas-ei2。图3C中是pKcas-ei1 (泳道6和7) 与pKcas-ei2(泳道8和9) 进行XbaⅠ和HindⅢ酶切的电泳图,均获得了符合预期大小的4.0 kb片段;泳道10和11利用Hist-F/R引物验证2种重组质粒中的4.0 kb的目的片段;利用sgRNA-1/R和sgRNA-2/R引物分别从pKcas-ei1和pKcas-ei2质粒中扩增出130 bp左右编码sgRNA的小片段。最后经过测序证实2种质粒构建成功。通过原生质体转化的方法导入到S.spiramyceticus1941中。
2.3 筛选出目的菌株
利用Am抗性筛选出含有pKcas-ei1和pKcas-ei2重组质粒的阳性转化子。经过Tsr诱导Cas9表达后,通过传代和在37 ℃培养使质粒停止复制,筛选出Am抗性消失的菌株 (图4),再利用PCR筛选出既消除质粒又发生同源双交换的目的菌株。只有pKcas-ei1的转化子筛选出了阳性菌株ΔEI,而含pKcas-ei2的转化子均未筛选出目的菌株。通过bsm4-LF/RR引物在原株S.spiramyceticus1941和ΔEI突变株中都扩增出了大约6 kb的特异性条带 (图3F),经过序列测定,证实在ΔEI突变株中目的基因bsm4确实被ermEp*-ist取代,而且其上下游基因也未发生突变。
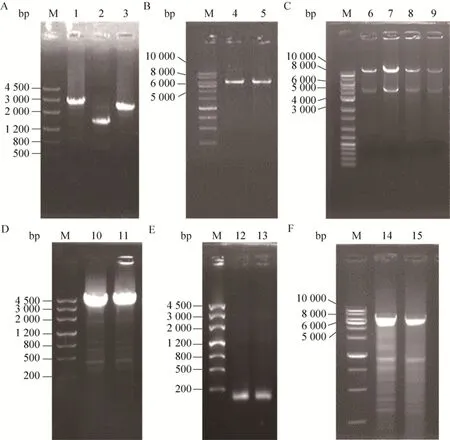
图3 三种重组质粒的电泳检测图Fig.3 Identification of the three recombinant plasmids by electrophoretic detection.1: left arm; 2: ermEp*-ist; 3: right arm; 4–5: pKC-ei (bsm4-LF/RR); 6–7: pKcas-ei1 (Hind Ⅲ/XbaⅠ); 8–9: pKcas-ei2 (Hind Ⅲ/XbaⅠ); 10: pKcas-ei1(Hist-F/R); 11: pKcas-ei2 (Hist-F/R); 12: pKcas-ei1 (sgRNA-1/R); 13: pKcas-ei2 (sgRNA-2/R); 14: S.spiramyceticus 1941; 15: ΔEI; M: DNA marker.
2.4 检测目的菌株ΔEI的发酵产物
通过HPLC检测S.spiramyceticus1941原株和ΔEI突变株的发酵产物,与原株 (图5B) 相比,在ΔEI突变株中没有保留时间与SP Ⅱ和SP Ⅲ组分相似的产物出现,只有产物a保留时间为4.749 min,与SPⅠ的保留时间和紫外吸收光谱一致;与BT标准品 (图5A) 对比,ΔEI中产物b的保留时间为16.596 min,与ISP Ⅰ的保留时间一致(图5C)。ΔEI突变株的发酵产物经一级质谱全扫描,并未发现ISP Ⅱ和Ⅲ组分分子量相同的化合物峰。在ΔEI突变株中化合物a的离子峰[M+H]+为m/z843.5,与SP Ⅰ一致;产物b的离子峰[M+H]+为m/z927.62 (图5D),与ISP Ⅰ的一致。而且产物b的二级碎片离子分别为768.36、699.39、540.37及402.28,也与ISP Ⅰ的裂解规律 (图6) 和碎片峰数据一致 (图5E和表2)。在ΔEI突变株的发酵产物中保留时间为26 min左右的峰为其他发酵产物,其[M+H]+为m/z279,远小于螺旋霉素的分子量。因此ΔEI突变株发酵产物中只有SP Ⅰ和ISP Ⅰ组分,证实新的ISP Ⅰ菌株构建成功。

图4 通过Am抗性和温度筛选出的目的菌株Fig.4 The target strains screened by Am resistance and temperature.
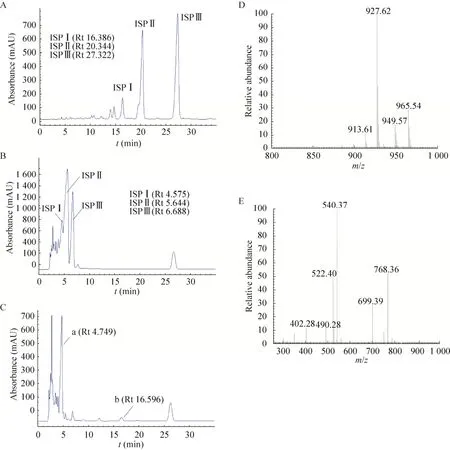
图5 ΔEI突变株发酵产物的高效液相检测图和质谱检测图Fig.5 The profiles of HPLC and MS for the fermentation products of ΔEI strain.(A–C) HPLC detection.A: BT; B:S.spiramyceticus 1941; C: ΔEI.(D) The MS spectrum of compound b in ΔEI strain.(E) Product ion spectrum of [M+H]+of compound b at m/z 927.
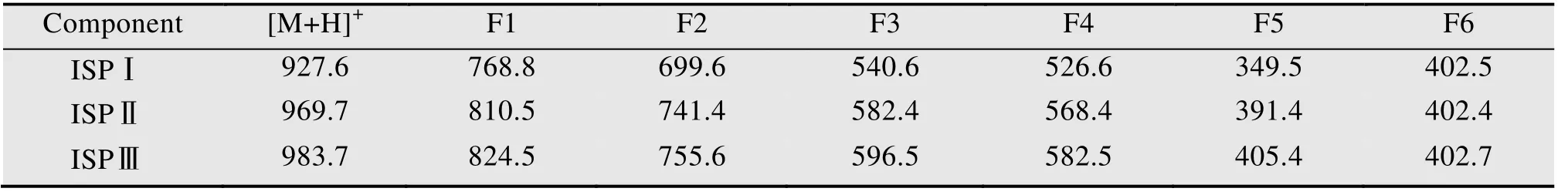
表2 异戊酰螺旋霉素的二级质谱碎片峰Table 2 The secondary MS data of isovalerylspiramycin
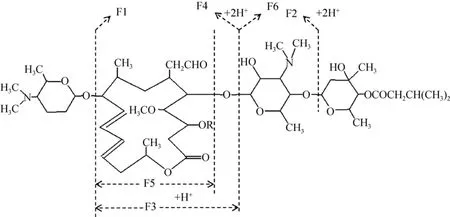
图6 异戊酰螺旋霉素离子的裂解规律图[16]Fig.6 The cleavage pattern of isovalerylspiramycin’s ion[16].
3 讨论
ISP Ⅰ是BT中的一个主组分,其抗菌活性与BT相似,但其发酵产物质控更容易,还可以做成注射剂型。本研究获得ISP Ⅰ新产生菌,只通过一次基因改造就实现目的基因的替换,而且不带有抗性标记,可以持续进行遗传操作来优化菌种。
实验前期选择经典同源重组的方法,通过替换质粒pKC-ei导入目的菌株进行同源双交换,因为没有抗性选择压力,自然发生同源双交换的概率比较低,很难筛选到目的菌株。CRISPR-Cas9作为一种新的基因编辑技术可以通过sgRNA指导Cas9蛋白完成对靶DNA的双链剪切而被广泛使用,近几年已经成功将该技术应用于腺病毒[17]、果蝇[18]、拟南芥[19]、水稻[20]、斑马鱼[21]等模式物种的基因修饰或改造中。在前期工作的基础上利用CRISPR-Cas9基因编辑系统完成了bsm4基因的替换。开始时将6.6 kb的外源片段均克隆至pKCcas9do质粒上,重组质粒总长度近20 kb,因质粒过大导致原生质体转化试验失败。缩短左右同源片段后,构建出的新质粒顺利导入S.spiramyceticus1941中。经过Tsr的诱导、Cas9的切割及同源片段的互补后,最终筛选出发生同源双交换的目的菌株。但只有pKcas-ei1筛选出了转化子,pKcas-ei2一直未能成功筛选出目的菌株,原因可能是第2个切割位点不太适合CRISPR-Cas9系统,还有可能是发生脱靶效应导致重要基因失活。
ermEp*是链霉菌中组成型强启动子,包含有2个-35区和-10区,属于多重启动子,可能还具有双方向调控转录的功能[22],在3位酰基转移酶基因的上游是23S rRNA的甲基转移酶基因,是一种修饰23S rRNA而获得抗生素抗性的基因,抗生素产生菌的产抗生素能力与其对抗生素的抗性之间具有一定的相关性,抗性基因的高表达通常能提高抗生素的产量[23–25]。用ermEp*-ist基因替换bsm4基因可以同时实现2个目标:1) 将3位酰基转移酶基因删除,菌株只产生SP Ⅰ;2) 高表达的ist基因整合到宿主染色体上稳定存在,而且位于螺旋霉素生物合成基因簇之中,有利于进行异戊酰基化修饰。另外,ermEp*还可能会提高上游的抗性基因的表达,从而增加螺旋霉素的产量。而且只经过1次遗传操作和含有1个ist基因,对菌株的影响较小,菌株的遗传稳定性更好。
从HPLC检测结果来看,尚有大量的SP Ⅰ没有转化成ISP Ⅰ,说明ermEp*控制下的ist基因并没有获得理想的高表达,但其效价为420 μg/mL,比原始的ISP Ⅰ菌株提高了2倍多。通常来讲,经过基因工程操作的菌株的产量开始时一般都偏低,还需要进行相应的育种分离操作,才能筛选出效价和ISP Ⅰ都较高的新菌株。将来还可利用CRISPR-Cas9系统把已经构建的ist高表达盒[26]导入到ΔEI菌株中,将会进一步提高SP Ⅰ转化为ISP Ⅰ的产量,还可进行其他遗传操作来获得更加优质高产ISP Ⅰ的菌株。
