先天性肾病综合征家系的临床表型和NPHS1基因突变分析
2019-03-26,,,,
, , ,,
(空军军医大学第一附属医院妇产科,陕西 西安 710032)
先天性肾病综合征(congenital nephrotic syndrome,CNS)是一类非常罕见的肾病综合征,通常定义为在出生后3个月内发生的肾病综合征,临床表现符合大量蛋白尿、低白蛋白血症、严重水肿和高胆固醇血症的肾病综合征的诊断标准。肾病综合征在儿童中的患病率为12/100 000~16/100 000儿童,年发病率为2/100 000~7/100 000儿童,其中CNS的发病更为罕见,大约为0.5/100 000新生儿[1]。根据病因CNS可为分原发性(遗传性) CNS和继发性(非遗传性)CNS。原发性CNS是肾小球滤过屏障组成蛋白的编码基因或其他相关基因突变所致,继发性CNS多因宫内感染或母亲疾病等所致,其中遗传性因素所致的CNS占主要地位。随着分子遗传学技术的不断发展,人们越来越重视对遗传性肾病基因层面的研究。本研究采用二代测序(next generation sequencing ,NGS)的方法对一个CNS家系患病胎儿进行基因突变检测,数据分析结果显示在该患儿的NPHS1基因发生复合杂合突变c.712+5G>A和c.3325C>T,分别遗传自其父母,且c.712+5G>A为新报道的突变位点。再结合PCR和Sanger测序在该家系中进行验证,从遗传学的角度明确了该家系中患儿CNS的发病原因,为其临床诊断提供了有力的证据,为该家系的遗传咨询和产前诊断提供了分子依据。
1资料与方法
1.1临床资料
1.1.1第一胎患儿临床资料
患儿,女,出生于2015年4月,3月龄时因CNS、肺部感染、多器官功能衰竭夭折。患儿母亲孕期无感冒、发热等感染史,无放射线及毒物接触史,无宠物接触史。当地医院孕期产检唐氏综合征筛查、胎儿系统B超及四维彩超均未见明显异常。该患儿胎龄34+4周因“横位、胎膜早破、绒毛膜羊膜炎”于当地医院剖宫产娩出。分娩情况:孕妇体温38.3℃,脉搏118次/分。剖宫产术前血常规:白细胞(WBC)13.47×1012/L,中性粒细胞百分比(N%)93.70%;C-反应蛋白(C-reactive potein,CRP)49.30mg/L。剖宫产术中见羊水黄绿色,量约300mL,胎盘明显增大,质地糟脆。以臀位娩出一女活婴,Apgar评分7-9-10分。新生儿身长46cm,体重2 220g。查体:新生儿生命体征平稳,面貌外观未见明显异常,肩背部、双下肢明显凹陷性水肿,四肢肌张力减低,左手桥贯掌,双侧小腿过度屈曲紧贴大腿,双下肢伸展受限。一胎患儿出生第1天、第8天、2月龄时尿常规及生化检验结果见表1。腹部超声:双肾实质光电稍增强,腹水形成。患儿因“早产儿、新生儿轻度窒息、CNS、低蛋白血症”于当地医院NICU住院治疗,给予暖箱保暖、氧帐吸氧、预防出血、抗感染、静脉营养、补充蛋白等对症支持治疗后,患儿全身水肿较前明显减轻,肌张力有所提升,进食尚可,好转出院。之后反复因“低蛋白血症、肺部感染”住院治疗,于3月龄时夭折。
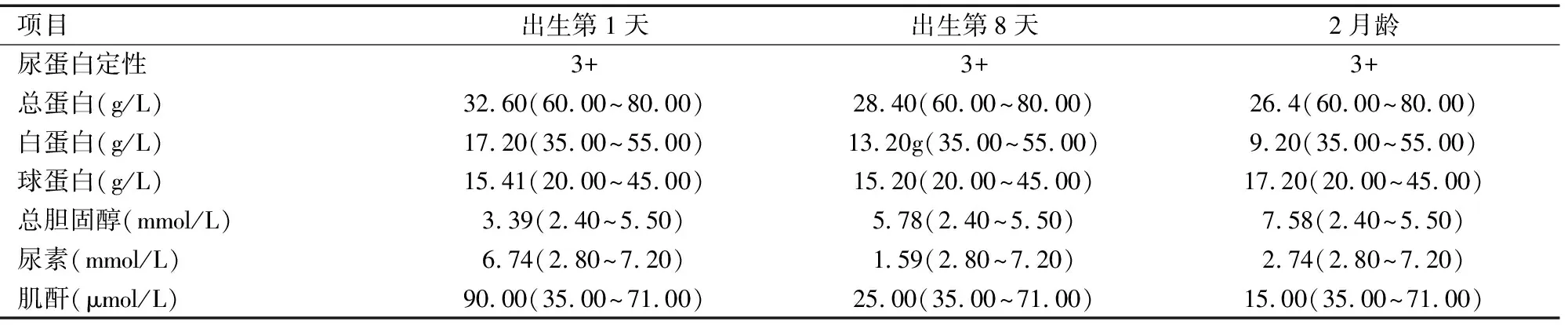
表1 一胎患儿尿常规及肝功能检测结果
注:括号中为参考范围。
1.1.2第二胎胎儿临床资料
二胎胎儿,因孕期产检发现胎儿CNS于2017年7月孕27周时在当地医院引产。该家系母亲孕前检查:血常规、尿常规、感染四项、优生四项、白带常规等均未见异常。孕期产检:孕早期胎儿颈项透明层(nuchal translucency,NT)值正常;唐氏筛查:神经管缺陷高风险;孕21周胎儿系统B超提示:臀位、胎儿双肾体积略增大,回声略增强、下腹部部分肠管回声增强、胎儿双心室强光点。夫妻双方染色体核型未见异常。因既往不良孕产史、唐氏筛查神经管缺陷高风险、超声软指标异常,经孕妇及家属知情同意后于孕22周行羊水穿刺,羊水穿刺结果:胎儿染色体核型:46,XX;染色体微阵列(CMA)检测结果阴性;羊水甲胎蛋白(AFP)>121 000ng/mL(正常参考值:1 266~26 300ng/mL);羊水血清胆碱酯酶200IU/L;羊水蛋白定性:4+;羊水微量蛋白8 780.00mg/L(正常参考值:<30.00mg/L);红细胞定性2+。与同孕周羊水AFP水平相比该胎儿AFP明显升高,羊水中大量蛋白尿,故高度疑诊为CNS。与该家系夫妻双方反复沟通后,其要求终止妊娠。
家族史:该家系夫妻双方平素均体健,否认近亲结婚,双方家族中无类似疾病患者,无遗传性疾病史。该CNS家系图见图1。
1.2方法
1.2.1基因组DNA提取
该CNS家系夫妻双方知情同意后,采用依地酸钠(ethylene diamine tetraacetic acid,EDTA)抗凝管抽取该夫妻双方静脉血各5mL,按血液基因组NPDNA提取试剂盒(北京天根生物科技有限公司)提取外周血基因组DNA,-20℃保存备用。
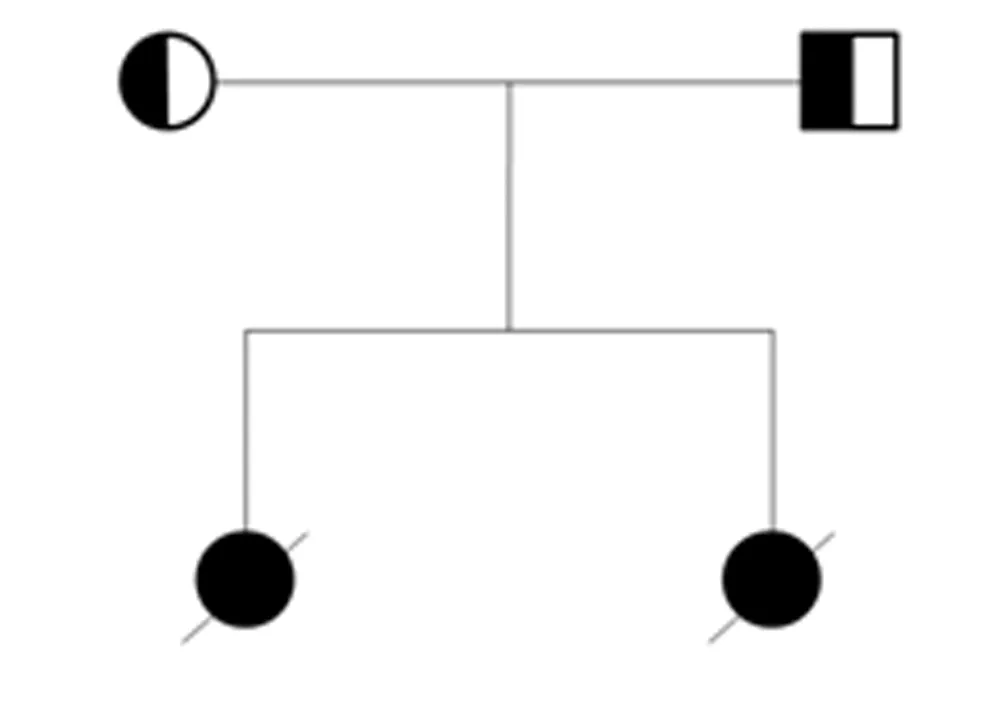
图1 先天性肾病综合征家系图
1.2.2二代测序
将该家系中的二胎患病胎儿的新鲜脐带组织5g左右,剔除结缔组织,吸水纸吸干血液,剪碎后放入研钵,倒入液氮磨成粉末,按DNA提取试剂盒(北京天根生物科技有限公司)说明书提取胎儿DNA,送至北京康旭医学检验所,利用目标序列捕获高通量测序技术,通过特异性基因捕获探针对多个肾病相关基因目标区域DNA片段进行捕获,并结合NGS进行测序分析。
1.2.3 PCR和Sanger测序验证
根据NGS的测序结果,用PCR结合Sanger测序对二胎胎儿及其父母进行突变检测和验证。采用Primer 5.0软件设计引物,扩增NPHS1基因的6号外显子和26号外显子序列。引物序列:NPHS1-6F:TTGGTCAGATGTGGGGCTCT,6R:ACTCG ̄ATGACAGGGGGTCCT;NPHS1-26F:AGGCT ̄CTGAGGGAGGTTTGG,26R:GATCAAGGCA ̄CCCAGTCCAG。引物由上海生工生物工程有限公司合成。PCR反应条件为:95℃10min,95℃30s,60℃30s,72℃45s,共35个循环,最后72℃延伸5min。PCR产物用1%琼脂糖凝胶电泳检测,之后进行双向测序。
2结果
2.1 NGS结果
分析NGS的数据发现,二胎胎儿NPHS1基因存在6号外显子c.712+5G>A及26号外显子c.3325C>T两个位点的复合杂合突变。变异c.712+5G>A的致病性尚未见文献报道,为剪切位点突变;c.3325C>T(编码区第3 325号核苷酸由C变为T),该变异导致编码第1 109号氨基酸Arg的密码子变为终止密码子(p.Arg1109Ter),从而使肽链合成提前终止,形成缺少了132个氨基酸的截短蛋白,为无义突变,见图2。c.3325C>T的致病性已经有文献报道,与CNS Finnish型相关。上述变异均不属于多态性变化,在人群中发生的频率极低(所参考数据库:1000Genomes、dbSNP)。
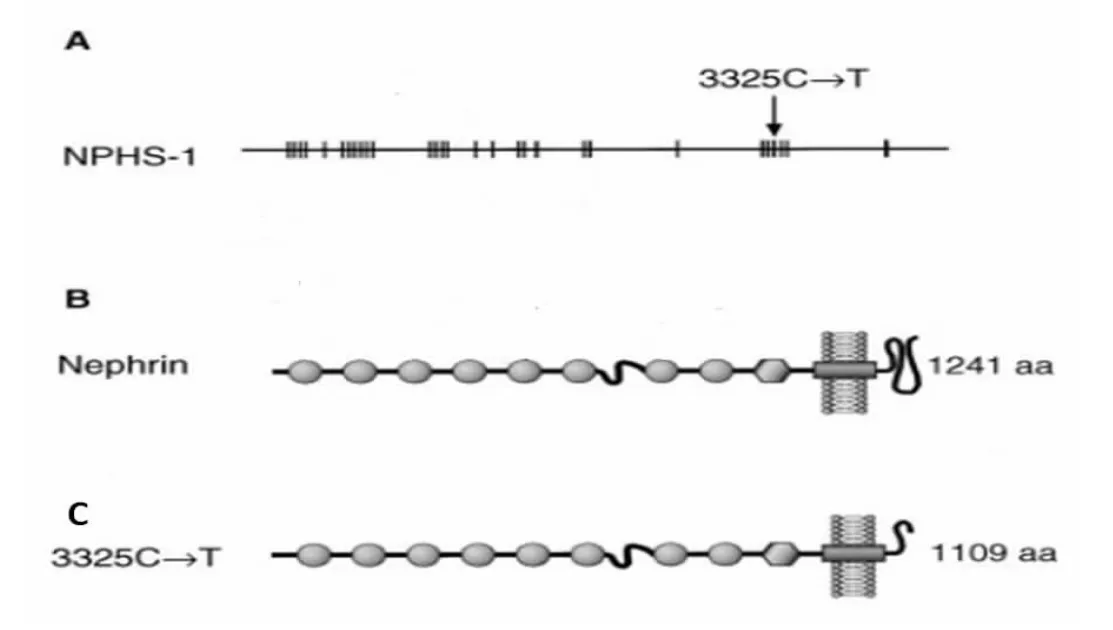
注:A.NPHS1基因;B.NPHS1基因编码的正常Nephrin蛋白;C.NPHS1基因c.3325C>T(p.Arg1109Ter)突变后形成的截短蛋白。
图2 NPHS1基因及Nephrin蛋白结构示意图
Fig.2 Structure diagram of NPHS1 and Nephrin
2.2 Sanger测序验证结果
对二胎胎儿及其父母用Sanger测序法验证上述位点的突变情况,证明胎儿位于第6号外显子的c.712+5G>A(编码区第712号核苷酸后内含子中第5位核苷酸由G变为A)变异来源于父亲;患儿位于第26号外显子c.3325C>T(编码区第3 325号核苷酸由C变为T)的核苷酸变异来源于母亲,与二代测序结果一致。其父母分别为上述杂合突变的携带者,该遗传方式符合常染色体隐性遗传,见图3。
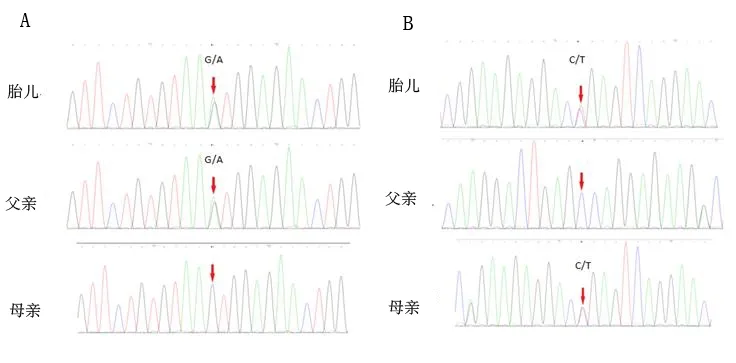
注:A为胎儿NPHS1基因c.712+5G>A变异来自于父亲;B为胎儿NPHS1基因c.3325C>T变异来自于母亲。红色箭头指示突变位点。
图3家系NPHS1基因Sanger测序验证结果
Fig.3 Sanger sequencing verification results of NPHS1
3讨论
3.1 CNS的基本概述
CNS是指生后3个月内即出现符合肾病综合征(大量蛋白尿、低蛋白血症、严重水肿和高脂血症)的临床表现。CNS的预后很差,大多数病例死于出生后的半年以内,有部分病例在孕期宫内就已经发病,甚至是胎死宫内,原因在于CNS往往表现为激素耐药故很快进展为急性肾衰竭,且合并感染等,唯一有效的治疗方式是肾移植[1]。以往多根据肾病综合征发生的早晚及严重程度进行分型,随着分子遗传学技术的快速发展,近些年不断有引起CNS的致病基因被定位、克隆,所以越来越多的学者倾向于根据致病基因进行分型。有报道称将近60%的新生儿肾病综合征患者是由于发生基因突变而致病的[2]。CNS可根据有无其他系统受累分为孤立性先天性肾病综合征和系统性先天性肾病综合征。NPHS1基因突变是孤立性CNS的常见致病原因,除此之外NPHS2、PLCE1、WT1、LAMB2、COQ2和LMX1B等基因突变也可致CNS。CNS芬兰型(CNF)为最常见的孤立性CNS,因编码Nephrin的NPHS1突变所致,为常染色体隐性遗传,在芬兰其发病率约为1/8 200,在芬兰以外的地区发病率明显低,我国仅有散发病例报道[3-5]。
3.2 CNS的常见临床表型
CNF临床表现为典型的肾病综合征,但其病因、病理变化、预后等与年长儿或成人不同,临床表型变异相对较小。CNF患儿出生时往往为早产,臀位、羊水污染和出生窒息史多见,出生体重1 500~3 500g,胎盘为大胎盘,胎盘重量可超过胎儿体重的25%,多无肾外畸形。患儿起病早,宫内即可有临床表现,出生后3个月之内出现蛋白尿、水肿。临床多见腹胀、腹水、矮小、耳低、眼距宽、鼻小、颅缝宽、骨龄和智力发育迟缓等,部分患儿可表现为肌张力低下[6]。肾脏组织活检可能出现肾小管特征性改变,即近曲小管囊性扩张,又被称为“小囊性病(microcystic disease)”,但并非见于所有患者,不是特征性的病理改变。实验室检查:尿常规见尿蛋白增加,4~13g/dL,血尿常见;血生化见血浆白蛋白<30g/L,血胆固醇:5.2~15.6mmol/L,血肌酐和尿素在病程早期正常,后期可增高。腹部B超部分可表现为肾脏增大、肾皮质高回声与髓质界限分明。自1970年以来,羊水中AFP浓度的升高被广泛用于CNF的产前诊断,胎儿血清中含有高浓度的AFP,由于CNS患儿在宫内排出蛋白尿,羊水的AFP水平显著增高,先天性神经管发育不全,也可出现羊水AFP水平增高,但其胆碱酯酶水平常同时增高可与CNF相鉴别。但有报道称NPHS1基因突变携带者也可出现羊水AFP浓度不同程度的升高,故羊水AFP浓度的升高不能作为CNF的特异性诊断指标,只能作为重要的参考指标。以上辅助检查均具有一定的辅助诊断价值,CNS的临床诊断主要根据肾病综合征的表现和发病年龄而定,分子诊断发现致病基因为诊断的金标准。回顾该家系的两次妊娠史发现,一胎患儿出生后即表现为典型的低蛋白血症、水肿、高脂血症、大量蛋白尿,且出生时为早产,伴有宫内感染、大胎盘,诊断为CNS;二胎胎儿宫内即表现为大量蛋白尿,羊水穿刺羊水中AFP浓度异常升高,CNS高度可疑。对二胎胎儿进行二代测序,检测到胎儿在NPHS1基因上携带父源性c.712+5G>A突变和母源性突变c.3325C>T,其中c.3325C>T为已报道的CNF的主要热点突变,c.712+5G>A突变为剪切位点突变,可能造成NPHS1基因转录水平剪接异常,从而导致编码的Nephrin蛋白结构或功能发生异常,进一步影响肾小球的滤过功能。
3.3 NPHS1基因的功能和相关致病性
NPHS1基因位于19q12~q13.1,拥有29个外显子,能转录4.3kb的mRNA,完整的cDNA编码含有1 421个氨基酸的足细胞结构分子Nephrin(肾病蛋白),是肾小球滤过屏障中的重要组成蛋白和功能蛋白。Nephrin蛋白属于免疫球蛋白超家族的细胞黏附分子,主要表达在肾脏,在肾小球滤过功能中发挥重要作用,是一种位于肾小球足突细胞裂孔膜的1型跨膜蛋白。Nephrin蛋白由胞外区、跨膜区和胞内区组成。胞外区包括8个免疫球蛋白(Ig)基序和1个纤连蛋白-Ⅲ样模块,NPHS1基因的2~20号外显子编码8个Ig基序,第22~23外显子编码纤连蛋白-Ⅲ样模块,24号外显子编码跨膜区,25~29号外显子编码胞内区。人NPHS1基因2号外显子的2个碱基对(CT)缺失(c.121delCT;p.L41fsX91)将导致Finmajor型CNF,而26号外显子的1个碱基对无义突变(c.3325C>T;p.R1109X)将导致Finminor型CNF。Finmajor型CNF约占芬兰CNS病例的78%,Finminor型CNF约占芬兰CNS病例的16%,然而这两类突变在其他种族和国家中并不多见。目前已报道的NPHS1基因突变约255种(突变类型多样,其中以错义突变最常见,其他还可见到无义突变、插入/缺失突变、剪切位点突变)[7]。本研究家系中NPHS1基因存在两种突变类型,其中来自于母亲的c.3325C>T变异为Finminor突变,为NPHS1基因26号外显子的无义突变,编码形成长1 109个氨基酸残基,缺少了Nephrin蛋白的胞内区(见图2);来自父亲的c.712+5G>A变异为剪切位点突变,在HGMD数据库、千人基因组数据库、dbSNP数据库等均未见报道,其是否致病需体外分子实验进一步验证,由于该家系两个患儿均未行尸检故无法进一步验证。但结合该家系的病史及CNF的遗传方式,高度怀疑c.712+5G>A变异为NPHS1的新发致病性突变。由此推测NPHS1基因的两个复合杂合位点突变是导致该CNS家系致病的病因。
3.4 CNS的治疗及预后
CNS的治疗比较棘手,需尽早明确疾病类型,区分出原发性CNS和继发性CNS,根据分型制定相应的治疗方案,继发性CNS针对病因行抗感染治疗、免疫抑制治疗等。遗传性CNS中激素耐药性CNS约占激素耐药型肾病综合征的10%~20%,其中家族性CNS糖皮质激素的耐药性更高,NPHS2基因突变导致的CNS又被称为激素耐药型CNS,但有报道称NPHS1导致的CNS预后要比NPHS2型肾病综合征更差,对激素和免疫抑制剂的抗性更强[8]。所以多数CNS对于糖皮质激素和免疫抑制剂治疗无效,肾移植是最佳选择。部分学者提出去肾脏治疗,可在生后6~10个月行双侧肾切除,透析维持3~4个月,保证患儿成长,改善营养状况,于2岁以后或体重达到7kg时进行肾移植[9]。而在病程早期主要是提供足够营养,纠正低蛋白血症,防止感染及电解质紊乱。
近年来NGS技术已经广泛应用于临床疾病的基因突变检测,研究者可以根据基因突变的特点与其他传统的检测技术结合使用,相互配合取长补短,帮助临床上更加经济、精准、快速地诊断疾病。CNS的致病基因种类繁多,每个基因的致病位动辄上百,如果用Sanger测序逐一排查经济效率极其低下,所以本研究选择NGS和Sanger测序相结合的方法进行基因诊断,帮助快速准确的找到致病基因及致病位点,大大提高了检测效率[10]。本研究进一步丰富了NPHS1基因突变数据库,为CNS的诊断、治疗提供了有力的分子依据,为该家系的遗传咨询和产前诊断提供了重要证据。
